
The Mitochondrial Switch: How Silencing MTCH2 Could Redefine Obesity Therapeutics
This week in the Guardrail, we explore groundbreaking research from the Weizmann Institute of Science on MTCH2—a mitochondrial target that could solve the industry's biggest GLP-1 challenge
By Michael Bronfman
August 3, 2026
The metabolic landscape has undergone a massive shift over the past few years. Incretin-based therapeutics, particularly GLP-1 receptor agonists and dual GLP-1 and GIP receptor agonists, have transformed how clinicians treat obesity and metabolic disease. Patients routinely see body weight reductions ranging from 15% to 25%, alongside improvements in glycemic control, cardiovascular risk profiles, and liver fat accumulation.
Yet, as clinical adoption expands, a significant challenge has emerged. Standard calorie restriction combined with incretin therapy causes loss of lean muscle tissue alongside fat mass. In many clinical trials, muscle loss accounts for anywhere between 25% and 40% of total weight reduced. For older adults, individuals with pre-existing sarcopenia, or patients undergoing long-term metabolic treatment, losing lean muscle mass poses genuine risks to metabolic rate, functional mobility, and overall longevity.
To solve this problem, researchers at the Weizmann Institute of Science directed their attention toward fundamental cellular energy dynamics. Their research, published in The EMBO Journal, explores Mitochondrial Carrier 2, a protein commonly known as MTCH2 or Mitch.
By silencing this single protein in human cells, scientists uncovered a cellular mechanism that increases energy expenditure while simultaneously stopping immature preadipocytes from transforming into mature, fat-storing cells.
What Is MTCH2 and Why Does It Matter?
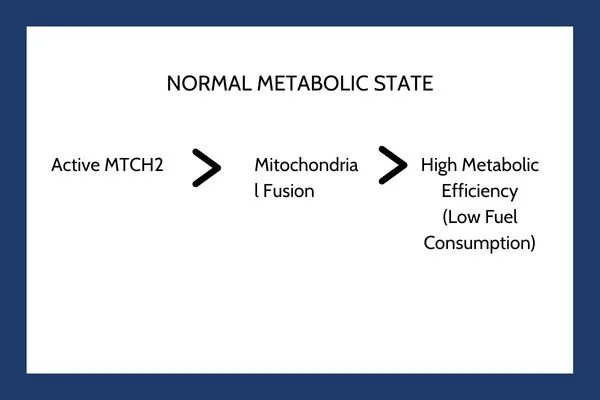
MTCH2 is an outer mitochondrial membrane protein that plays a key role in regulating mitochondrial dynamics, lipid transport, and apoptotic signaling pathways. Mitochondria are constantly changing structures. They fuse together into connected networks or divide into smaller, individual units through a process known as mitochondrial dynamics.
Under normal physiological conditions, MTCH2 acts as a metabolic gatekeeper. It supports mitochondrial fusion and maintains organelle stability. When mitochondria fuse into dense networks, they operate at peak efficiency, generating adenosine triphosphate through oxidative phosphorylation while minimizing energy wastage.
When researchers generated mouse models lacking MTCH2 in previous studies, the animals demonstrated a striking phenotype. They remained lean even when fed high fat diets and exhibited remarkable physical endurance. Translating these findings from animal models to human cellular biology remained a critical milestone.
The Mechanism: Mitochondrial Fission and Energy Demand
To evaluate whether this phenomenon holds true in humans, the research team used genetic engineering techniques to knock down or silence MTCH2 in human cell lines. The outcome was immediate and distinct.
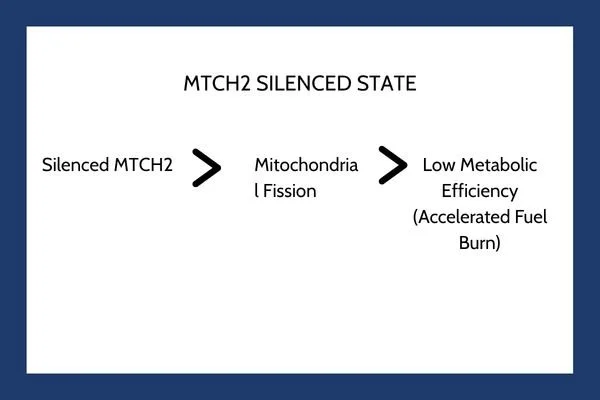
Without MTCH2, the mitochondrial network loses its ability to fuse efficiently. Instead, the mitochondria separate into smaller, fragmented organelles through fission. This structural alteration significantly impacts cellular bioenergetics.
When mitochondria shift into a fragmented state, their coupling efficiency drops. They produce less ATP per unit of nutrient substrate consumed. To compensate for this sudden energy drop and maintain basic cellular functions, the cell undergoes a energetic scramble. It ramps up metabolic flux, consuming stored triglycerides, fatty acids, and glucose at an accelerated rate.
Key Takeaway: Silencing MTCH2 forces human cells into a continuous state of high metabolic demand. Rather than storing excess nutrients as triglycerides, the cell burns through available fuel sources just to maintain basic energetic equilibrium.
This forced energy expenditure occurs independently of classical uncoupling proteins like UCP1, which drives thermogenesis in brown adipose tissue. Instead, MTCH2 silencing fundamentally alters structural energy transfer within the outer mitochondrial membrane.
Blocking Adipogenesis: Stopping Fat Cells at the Source
The acceleration of metabolic rate is only one half of the story. The study demonstrated a second critical effect: blocking adipogenesis.
Adipogenesis is the multi step differentiation pathway through which precursor cells evolve into mature, lipid laden adipocytes. During normal adipose tissue expansion, precursor cells take up fatty acids, upregulate key transcription factors like PPAR gamma and C/EBP alpha, and accumulate large lipid droplets.
When the researchers silenced MTCH2 in human preadipocytes, the differentiation cascade halted. The cells could no longer execute the metabolic reprograming necessary to build lipid storage vessels.

By preventing precursor cells from expanding into fully mature adipocytes, MTCH2 inhibition creates cellular resistance against fat storage. Excess carbohydrates and lipids circulating in the extracellular environment are either oxidized immediately to meet elevated baseline mitochondrial demand or cleared through alternative metabolic pathways.
Comparing Treatment Modalities: Incretins vs. MTCH2 Targets
Understanding where MTCH2 targeted mechanisms fit into the current obesity treatment landscape requires comparing them directly with established incretin therapies.
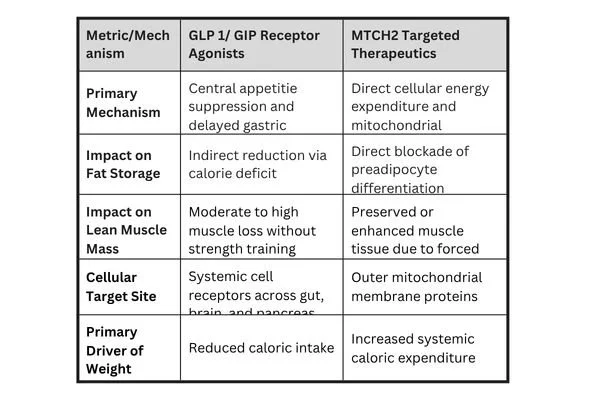
Current GLP-1 therapies act primarily through the central nervous system to reduce hunger and slow digestion, as documented in clinical resources on NCBI PubMed. While effective at reducing overall food intake, a calorie deficit created purely through reduced consumption causes the body to break down both adipose tissue and skeletal muscle protein for gluconeogenesis.
In contrast, an MTCH2-targeted mechanism acts directly at the organelle level. Because the cell demands higher energy flux to sustain itself, skeletal muscle tissue, which is dense with mitochondria, retains its structural requirement for fuel. Animal models lacking MTCH2 exhibited increased endurance capacity, suggesting that mitochondrial fragmentation in muscle tissue encourages fatty acid oxidation without promoting tissue atrophy.
Addressing the Lean Muscle Loss Dilemma
Preserving skeletal muscle during weight reduction is one of the most pressing goals in modern pharmaceutical development. Skeletal muscle is not simply a structural tissue; it is a major site of insulin-mediated glucose disposal, amino acid storage, and basal metabolic output.
When patients lose significant muscle mass during rapid weight loss:
Resting metabolic rate drops sharply, increasing the likelihood of weight regain once therapy stops.
Insulin sensitivity gains can plateau, as muscle tissue accounts for up to 80% of postprandial glucose uptake.
Physical strength and bone mineral density decrease, raising long-term cardiovascular and musculoskeletal risks.
By shifting the primary therapeutic driver from overall caloric restriction to targeted cellular energy expenditure, MTCH2 pathways offer a potential strategy for selective lipid reduction. Because the mechanism directly blocks adipogenesis while driving lipid burning within existing cells, it spares structural proteins needed for skeletal muscle integrity.
Challenges and Considerations for Drug Development
Translating cellular discovery into safe, orally bioavailable or injectable therapeutics presents significant pharmacological challenges. Because MTCH2 is expressed across multiple tissue types, drug developers must address several critical factors:
1. Tissue Specificity and Delivery Mechanisms
MTCH2 exists in cardiac, hepatic, renal, and central nervous system tissues. Inducing indiscriminate mitochondrial fragmentation across all organs could disrupt tissue function, particularly in high-demand organs like the heart. Future therapeutic strategies may require targeted delivery vehicles, such as antibody drug conjugates or lipid nanoparticles designed specifically for white adipose tissue and skeletal muscle.
2. Small Molecule Modulation versus Complete Knockdown
In genetic engineering experiments, researchers completely silence or knock out the target gene. In clinical medicine, total inhibition of an outer mitochondrial membrane protein may trigger unwanted cellular stress or apoptotic cascades over time. Drug discovery efforts will likely focus on allosteric modulators that partially reduce MTCH2 activity or disrupt specific protein-protein interactions without abolishing mitochondrial stability entirely.
3. Monitoring Systemic Biomarkers
Increasing baseline energy expenditure generates metabolic byproducts, heat, and reactive oxygen species. Clinical protocols evaluating MTCH2 modulators will need to monitor serum lactate levels, systemic inflammatory markers, mitochondrial health indicators, and body temperature regulation during early-phase safety trials.
Future Outlook for Next Generation Anti Obesity Therapeutics
The identification of MTCH2 as an energy expenditure switch highlights a fundamental shift in metabolic research. While first-generation anti-obesity drugs focused heavily on central nervous system appetite regulation, next-generation approaches aim to directly modify cellular bioenergetics and tissue composition.
Combinational approaches may represent the future of metabolic medicine. Combining a low-dose GLP-1 receptor agonist to manage appetite with a tissue-targeted MTCH2 modulator could allow clinicians to achieve profound lipid reduction while entirely preventing lean muscle loss.
If your organization is ready for the next frontier of metabolic drug development a. to navigate complex R&D landscapes, with precision strategy and deep scientific foresight, contact Metis Consulting Services today

The Grand Convergence of Compliance and Cutting-Edge Science
How the intersection of strict regulatory enforcement and breakthrough molecular science is redefining the modern pharmaceutical business model. This week in the Guardrail, we discuss The Grand Convergence of Compliance and Cutting-Edge Science

How the intersection of strict regulatory enforcement and breakthrough molecular science is redefining the modern pharmaceutical business model. This week in the Guardrail…
By Michael Bronfman
July 27, 2026
The global pharmaceutical landscape is undergoing a massive shift. For decades, drug discovery and commercial operations existed in separate silos. Scientists designed molecules in quiet laboratories, and supply chain managers built delivery pipelines in isolation, while legal teams handled regulatory compliance only after clinical data emerged. That disconnected era is officially over. Today, regulatory strategy, end-to-end tracing, and molecular design are merging into a single interconnected science.
As we progress through twenty twenty-six, developers face a dual challenge. They must navigate some of the strictest operational enforcement mandates in history while simultaneously scaling highly complex, personalized therapies that do not fit traditional manufacturing or licensing models. From the sudden end of drug tracing exemptions in the United States to clinical trials that target chronic diseases with “one-and-done " genetic edits, the rules of commercialization are being rewritten. The organizations finding success in this landscape are those that treat supply chain resilience and local regulatory intelligence not as administrative hurdles, but as core elements of their research and development strategy.
Supply Chain Risk and Serialization: The End of DSCSA Exemptions
In the United States, the safety net of regulatory grace periods has officially vanished. The Drug Supply Chain Security Act, a long-standing federal effort to protect patients by creating an interoperable electronic tracing system for prescription drugs, has entered its final era of absolute enforcement. The phased rollout reached a critical milestone when exemptions for manufacturers and repackagers expired in May 2025, followed closely by the expiration of wholesale distributor exemptions in August 2025.
The final transition occurred when dispenser exemptions for larger organizations ended in November 2025, pushing the entire domestic market into full unit-level serialization. Today, every single trading partner must possess the infrastructure to electronically trace, verify, and exchange transaction data at the individual package level. A helpful breakdown of these hard boundaries and the necessary internal tracking milestones is here: Intelliguard DSCSA Compliance Guide.
This complete transition to unit-level serialization has exposed a deep vulnerability in global logistics. The modern pharmaceutical pipeline is incredibly fragile. A single supplier failure, raw material bottleneck, or cold chain temperature excursion can instantly halt clinical progression. Under the current strict rules, the stakes are even higher. If physical drug packages arrive at a distributor or pharmacy but their accompanying digital data contains a clerical error, the entire shipment must be quarantined.
Even physically perfect, safe medicines are routinely blocked from distribution because of minor electronic data mismatches. For a detailed look at how organizations must update their standard operating procedures to handle these digital bottlenecks, the Two Labs DSCSA Exception Handling Analysis highlights the operational necessity of treating serialization data as a product-critical asset.
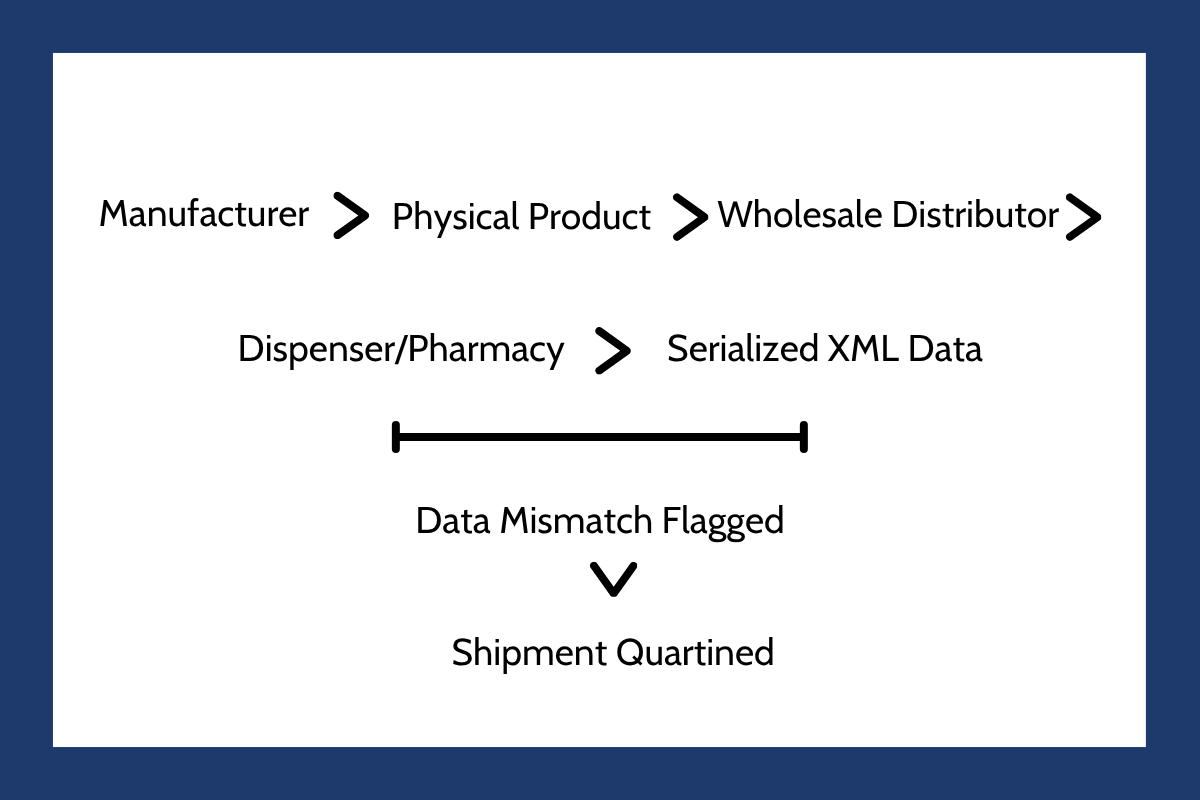
Because of these tight restrictions, biotech firms advancing candidates through clinical trials must construct highly resilient logistics networks years before submitting a Biologics License Application or New Drug Application. If a developer cannot guarantee the integrity of their data or the security of their active pharmaceutical ingredients, they risk sudden clinical holds or devastating launch delays. Supply chain resilience is no longer a post-approval operational concern; it is a primary metric that regulators evaluate during initial product reviews.
Base Editing and the Dawn of One and Done Chronic Disease Therapies
While compliance teams secure the physical supply chain, geneticists are shifting the boundaries of what is possible in preventative medicine. For years, the scientific community viewed genomic medicine through the lens of oncology and ultra-rare genetic mutations. However, a major paradigm shift is occurring as base editing moves into the domain of common, chronic diseases that affect millions of people globally.
Unlike early gene editing systems that rely on double-stranded DNA breaks, base editing allows scientists to make single-letter chemical transitions in a highly targeted, precise manner. This technology provides an incredibly clean way to silence specific disease-causing genes without triggering the cellular damage associated with double-stranded cuts.
The commercial reality of this science became undeniable following the landmark acquisition of Verve Therapeutics by Eli Lilly. At the center of this momentum is the base editing candidate known as VERVE one-hundred-two. Designed to permanently reduce low-density lipoprotein cholesterol, this therapeutic candidate has entered expanded Phase two clinical trials. The medicine functions by targeting and durably inactivating the PCSK9 gene directly within the human liver.
According to clinical updates published in the Eli Lilly PCSK9 Base Editor Press Release, a single infusion of the treatment achieved highly promising results, demonstrating an 88% reduction in PCSK9 protein levels and a sustained 62% drop in low-density lipoprotein cholesterol. This approach challenges the entire business model of chronic disease management. Historically, conditions like hyperlipidemia, hypertension, and cardiovascular disease required decades of daily oral pills or monthly subcutaneous injections. Compliance was notoriously poor, as patients frequently forgot doses or lost access to insurance. Transitioning to a “one-and-done genetic cure completely eliminates noncompliance.
However, it also presents an unprecedented challenge for drug developers, who must figure out how to manufacture, distribute, and price a single-dose therapy that permanently cures a common chronic disease. The manufacturing process relies heavily on lipid nanoparticles, requiring incredibly specialized raw materials that must be tracked under the newly enforced US serialization rules.
Epigenome Editing: The Next Reversible Frontier
As base editing demonstrates clinical success, another groundbreaking platform is rising to prominence. Epigenome editing is quickly emerging as the next major boundary beyond traditional CRISPR and base editing systems. While existing technologies focus on rewriting the physical letters of the genetic code, epigenome editing leaves the underlying DNA sequence completely untouched. Instead, it targets the molecular tags and histone modifications that dictate whether a gene is actively turned on or shut off.
This subtle approach is highly attractive to both drug developers and regulatory agencies because of one major feature: reversibility. Because the physical DNA sequence is never cut or permanently altered, the risk of permanent, dangerous off-target mutations is dramatically minimized. If an unexpected side effect occurs, or if a patient's biological requirements change over time, epigenetic modifications can theoretically be adjusted or reversed.
Pioneering biotechnology companies like nChroma Bio are actively developing proprietary epigenetic platforms to silence disease-causing proteins in real time. Because these therapies rely on complex post-translational modifications, their success hinges on extremely precise cellular delivery systems. Developers are investing heavily in customized chemical vectors that can reliably locate target tissues, dial down aberrant gene expression, and then safely exit the patient's system. This science offers a flexible middle ground between traditional small molecule drugs and permanent genetic modifications, establishing a highly versatile therapeutic class.
CAR-T Beyond Cancer: Resetting the Autoimmune System
The concept of cellular reprogramming is also sparking a major revolution within immunology. Chimeric antigen receptor T cell therapies, which were once reserved exclusively for terminal blood cancers, are proving to be exceptionally powerful tools for treating severe autoimmune disorders.
Under normal circumstances, autoimmune diseases like systemic lupus erythematosus and systemic sclerosis are managed with chronic immunosuppressive drugs. These conventional therapies do not cure the underlying disease; they merely suppress the entire immune system, leaving patients highly vulnerable to severe infections.
This dynamic is shifting rapidly. Early clinical data from trials running throughout 2026 have confirmed that CD19-targeted CAR-T therapies can achieve deep, durable remission in patients with severe autoimmune conditions. By collecting a patient's own immune cells, engineering them to target the CD19 antigen on aberrant B cells, and reintroducing them, clinicians can completely eliminate the autoantibody-producing cells responsible for the disease. Once these problematic B cells are cleared, the bone marrow naturally produces a fresh, healthy population of immune cells, effectively resetting the patient's immune system.
The clinical data supporting this profound shift has been highlighted by researchers globally. During presentations at the EULAR 2026 Congress CAR T Research Panel, clinical trial data demonstrated that dual target cell therapies could successfully clear skin fibrosis and stabilize progressive lung damage in systemic sclerosis patients. The safety profile of these therapies in autoimmune populations has also proven to be superior to that seen in oncology. Because the overall tumor burden is absent, the severe side effects that often complicate cancer treatments, such as severe cytokine release syndrome, are rarely observed in autoimmune cohorts.
This clinical expansion has triggered a scramble among biopharmaceutical companies to secure manufacturing capacity. Unlike oncology products, which are often produced in highly centralized facilities, autoimmune treatments require rapid, local delivery models to ensure that autologous cell products can be harvested, engineered, and returned to patients without operational delays.
Bespoke and Personalized Gene Editing: Baby KJ and N of 1 Paradigms
Perhaps the most inspiring and challenging frontier in modern medicine is the rise of bespoke, personalized gene editing. For decades, the pharmaceutical industry operated on a high-volume model, designing single therapies for millions of identical patients. However, for individuals suffering from ultra-rare genetic mutations, no commercial market existed. Developing a traditional drug for a population of one was simply too expensive and logistically impossible.
That paradigm changed forever with the historic treatment of an infant known as Baby KJ. Born with a lethal genetic condition that prevents the body from clearing toxic ammonia, called carbamoyl phosphate synthetase deficiency, his survival was highly unlikely.
In an unprecedented collaboration, researchers at the Children's Hospital of Philadelphia and the University of Pennsylvania engineered a customized, first-of-its-kind base editing therapy designed to correct the exact, single-point mutation in his DNA. The timeline of this historic medical achievement is detailed in The Future of Personalized Medicine: KJ's Story.
The regulatory response was equally historic. Recognizing the immediate threat to the child's life, the Food and Drug Administration reviewed the investigational application on an accelerated timeline, granting clearance in just one week. The success of this custom therapy has proven the technical feasibility of “N of 1 medicine,” opening a brand new avenue for patients with highly specific genetic conditions. An analytical review of this rapid development pipeline is featured on the Friends of Cancer Research Personalized Medicine Portal.
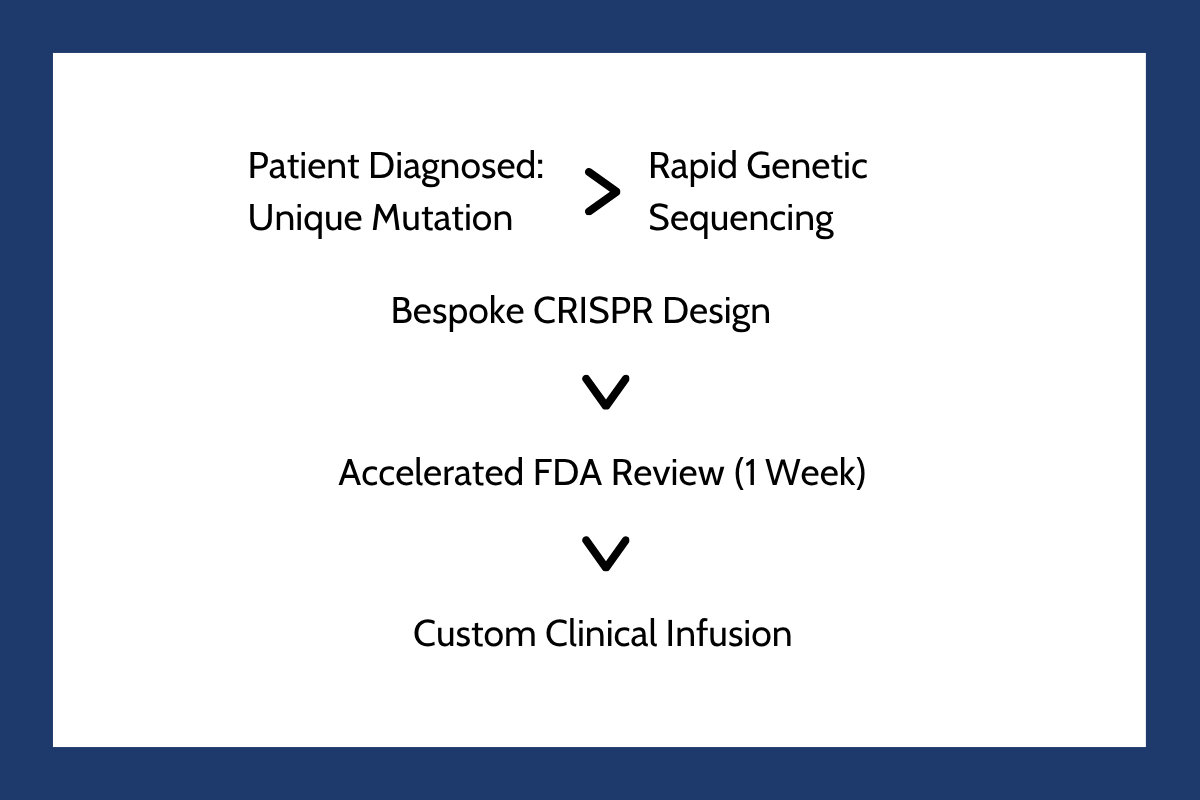
However, scaling these bespoke treatments remains an immense challenge. Traditional drug licensing and safety evaluation protocols are built around large-scale, multicenter trials with thousands of participants. A personalized, “N of 1” therapy cannot undergo standard Phase three clinical testing because there are no other patients to enroll.
To prevent these life-saving scientific breakthroughs from becoming stalled in regulatory paperwork, regulatory bodies must adopt highly flexible, platform-based approval pathways. Under this proposed model, regulators would evaluate and approve the editing tool and delivery system itself, allowing clinicians to simply swap out the genetic guide RNA sequence to match an individual patient's unique mutation.
Balancing Agile Operations with Unprecedented Medical Innovation
As we move forward through this year and beyond, the pharmaceutical sector finds itself at an incredible crossroads. The scientific capabilities of gene editing, epigenetics, and cellular reprogramming are advancing faster than ever before. Yet, the physical and electronic pathways required to deliver these miracles to patients have never been more tightly regulated or logistically complex.
Whether an organization is scaling a major cardiac base editor to treat millions of patients, or engineering a custom cell therapy to cure a single child, success requires deep operational agility. By integrating regulatory compliance directly into the earliest phases of molecular research, managing supply chain data as a critical asset, and building flexible manufacturing models, the global medical community can ensure that these historic discoveries successfully make their way from the laboratory bench to the patient bedside.
Don't let complex serialization mandates or strict regulatory hurdles delay your life-saving therapies. Contact Metis Consulting Services today to build flexible, bulletproof quality operations that keep your cutting-edge science moving seamlessly from lab to patient.

Outsourcing Oversight: FDA Form 483
Learn why maintaining rigorous third-party quality management is just as vital to your drug pipeline as the science itself. Read about why the critical operational risks highlighted in the FDA Form 483 Warning Letter are important to be aware of, and what dangerous oversight gaps exist in pharmaceutical outsourcing.

By Michael Bronfman
July 20, 2026
This week, discover why maintaining rigorous third-party quality management is just as vital to your drug pipeline as the science itself. Unpack with the "Guard Rail" the critical operational risks highlighted by FDA Form 483 citations and the dangerous oversight gaps in pharmaceutical outsourcing.
Imagine you are running a business that makes life-saving medicines. Because your business is growing fast, you decide to hire outside partners to manufacture the raw chemical ingredients, print the medical labels, and package the final pills. You sign the contracts, assuming these expert partners will handle everything perfectly.
Months later, an official investigator walks into your office and hands you a strict warning paper. The investigator found that your outside partner was using dirty equipment, and because you are the brand owner, your company is legally responsible.
This nightmare scenario happens constantly in the biotech world. The warning paper is called an FDA Form 483, which is the official document issued by investigators when they notice serious violations during factory visits.
A major blind spot in medicine manufacturing involves supplier quality management. Many biotech brands fail to realize that outsourcing production does not mean outsourcing legal liability. When a company fails to watch its external partners closely, it creates an oversight gap.
This gap frequently results in regulatory penalties, frozen pipelines, and missing medicines. Managing supplier risk is no longer just a minor paperwork requirement handled by a background department; it is a critical strategy needed to keep a pharmaceutical pipeline alive.
What is an FDA Form 483 and Why Does It Matter
To understand how this oversight gap happens, you have to understand how government monitoring works. The United States Food and Drug Administration conducts routine, unannounced physical inspections of factories where medicines are created. At the end of an inspection, if the investigator finds problems, they issue a Form 483 list of observations.
A Form 483 is not a final legal punishment, but it is a serious warning. If a business fails to fix the listed problems quickly, the situation can escalate into an official Warning Letter, which can shut down production entirely. You can read about the exact rules for these notices directly on the Inspection Observations Page of the FDA Website. This resource explains how field investigators evaluate factory conditions to ensure public safety.
In recent years, an increasing number of these forms point directly to poor supplier oversight. Government investigators are realizing that while biotech brands claim their products are pure, the brands rarely visit the third-party providers who supply the primary chemical components. This lack of visibility triggers immediate red flags during safety audits.
The Danger of Inadequate Supplier Qualification
The first place the oversight gap appears is during the initial hiring process, known as supplier qualification. Before a pharmaceutical firm buys a single gram of raw material from a third-party provider, they are required by law to run a deep background check on that supplier. This check ensures the provider follows current good manufacturing practices, which are the official quality rules for medicine creation.

Too often, growing biotech firms take shortcuts during this stage. Instead of sending a trained inspector to physically walk through a provider's facility, a firm might simply send a basic text questionnaire through email. They ask the provider if their facilities are clean, the provider checks a box saying yes, and the firm grants them approved status.
This careless approach is called paper qualification, and it fails to satisfy modern regulators. A paper survey cannot spot broken air ventilation systems, leaking pipes, or untrained factory workers. When government inspectors find out that a pharmaceutical company approved a critical material provider based solely on an emailed questionnaire, they issue a severe citation on a Form 483.
Insufficient Oversight of Contract Manufacturers
As biotech companies discover complex new therapies, they rely heavily on specialized partners called Contract Development and Manufacturing Organizations. These partners act as hired factories, taking a drug design from a small startup and mass-producing it inside large industrial facilities.
While outsourcing production makes financial sense, it frequently causes a breakdown in quality control. The biotech brand often adopts a hands-off attitude, believing the hired manufacturer is entirely responsible for daily safety. This is a massive mistake. Under international health frameworks, the brand owner always retains ultimate responsibility for the purity of the drug.
The official global expectations for this relationship are outlined in detail within the FDA Guidance Document on the Q10 Pharmaceutical Quality System. This international framework states that a comprehensive quality management system must extend to the control and review of outsourced activities.
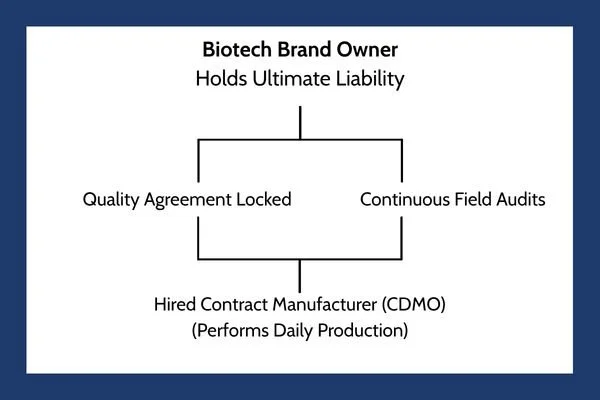
To eliminate the oversight gap, a biotech firm must treat their hired manufacturer as an extension of their own company. This integration requires:
Clear Quality Agreements: A legally binding document that states exactly which company handles specific testing steps, who approves batches, and how mistakes are corrected.
Continuous Person-in-Plant Oversight: Sending a full-time quality expert from the biotech brand to live at the hired manufacturer's facility, monitoring the production lines in person as your drug is made.
Shared Quality Data Tracking: Utilizing unified digital networks so the brand owner can review batch records, laboratory errors, and clean room data in real time, rather than waiting weeks for a text summary report.
The Hidden Risk of Incomplete Supplier Change Notifications
The third area where the oversight gap creates havoc involves supplier change notification processes. Medicine manufacturing is an incredibly delicate science. A microscopic shift in the raw materials can change how a drug behaves inside the human body, altering how fast a pill dissolves or causing unexpected allergic reactions.
Because the science is so sensitive, third-party providers are legally required to inform the biotech brand before making any modifications to their own facilities, raw materials, or testing tools. If a chemical supplier switches to a new raw material provider, or if a label printer updates their machine software, they must submit a formal change notification to the pharmaceutical brand owner.
Unfortunately, communication channels across these supply networks are often broken. A raw material provider might update a chemical purification tool, thinking the change is too minor to mention. If the pharmaceutical brand does not have a strict, mandatory system for tracking and forcing these updates, they remain completely unaware of the alteration.
When a government inspector reviews a biotech company's records and discovers that a raw chemical ingredient was altered without a formal evaluation, the factory receives an immediate Form 483 observation. The brand owner must prove they evaluated every single modification to ensure it did not alter the safety, identity, strength, quality, or purity of the distributed medicine.
Why Quality Management is a Major Pipeline Risk
When a pharmaceutical brand treats supplier oversight as an annoying checklist item rather than a core survival skill, they put their entire business pipeline at risk. The financial and operational damage caused by a single bad supplier can ruin years of scientific research.
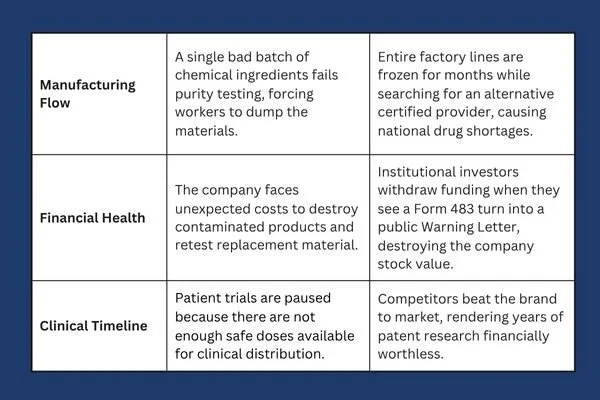
This table shows why supply chain monitoring is a critical commercial necessity. If your external partner fails a government inspection, your entire product launch can stall out, allowing rival businesses to capture the market.
Building a Modern Vendor Review Program
Fixing the oversight gap requires a complete shift in corporate mindset. Biotech organizations must stop choosing their partners based solely on the lowest price or the fastest timeline. Instead, they must construct an aggressive, data-driven vendor review program that treats safety as an active investment.
A modern vendor review strategy replaces casual trust with continuous verification. This process starts by ranking every partner based on risk. A vendor supplying the active chemical ingredient for an injection receives the highest risk rating and requires constant in-person monitoring and quarterly laboratory audits. A vendor supplying external cardboard shipping boxes receives a lower risk ranking, requiring fewer document checks.
Furthermore, companies must establish clear corrective and preventive action programs with their partners. If an external manufacturer makes a mistake, the biotech brand cannot simply accept a brief apology. The brand must force the partner to run a deep root cause analysis to discover why the error happened and prove that systemic changes were implemented to prevent the mistake from ever happening again.
Balancing Rapid Innovation with Supply Security
The biotech world is moving at a breathtaking pace, creating gene therapies and personalized medicines that were science fiction a decade ago. To bring these innovations to reality, companies must rely on a vast web of global suppliers. This interconnected network brings incredible strength, but it introduces immense vulnerability.
The persistent stream of Form 483 citations issued by health authorities is a clear warning that the industry is moving too fast for its own safety infrastructure. Designing an incredible molecule is meaningless if the third-party partner you hire to build it contaminates the batch with residue from a dirty machine.
As we look through 2026, the businesses that survive will be the ones that bridge the oversight gap completely. By treating supplier qualification, active partner communication, and strict change controls as vital priorities, biotech innovators protect their corporate reputation, satisfy tough regulators, and ensure a steady flow of safe therapies reaches the patients who depend on them.
Don't let your outsourcing strategy put your entire pipeline at risk: an unexpected FDA Form 483 or a partner's compliance oversight can derail years of clinical breakthroughs and market value. Contact Metis Consulting Services and establish an ironclad vendor review program, implement excellent quality agreements, and secure your global supply chain against regulatory friction.

European Union Pharmaceutical Industry Package
In December 2025, negotiators reached a historic political agreement on a major overhaul of drug laws known as the EU pharma package. As these massive new rules roll out across Europe, pharmaceutical companies face a complicated double reality. They must adapt to a unified European market while simultaneously managing messy, localized differences from country-specific ethics boards and regional testing requirements.

This week in the Guardrail, we dive into the sweeping changes brought by the landmark EU pharma package agreement and its impact on global drug development. Learn how balancing unified European regulations with intricate local hurdles is redefining pharmaceutical strategy as we advance through 2026.
By Michael Bronfman
July 13, 2026
Imagine trying to launch a new smartphone, but every single city you sell it in has a completely different rule about how the battery must work, how the screen must be tested, and what information needs to be printed on the box. You would spend all your time filling out paperwork instead of actually building great technology.
This exact challenge happens every day in the medical world. It is known as global regulatory divergence, which is a fancy way of saying that different countries have vastly different laws for approving and monitoring new life-saving medicines.
For decades, international organizations have tried to create global regulatory harmonization, which means getting health authorities across the globe to agree on a single, shared rulebook. When rules match, safe medicines can reach sick patients much faster.
However, achieving this balance is an ongoing tug-of-war. A massive example of this shift is happening right now in Europe. In December 2025, negotiators reached a historic political agreement on a major overhaul of drug laws known as the EU pharma package. As these massive new rules roll out across Europe, pharmaceutical companies face a complicated double reality. They must adapt to a unified European market while simultaneously managing messy, localized differences from country-specific ethics boards and regional testing requirements.
The EU Pharma Package: A Balanced Approach to Innovation
The newly agreed-upon European rules represent the most significant update to continental drug laws in over twenty years. The policy balances two urgent goals: getting critical treatments to patients much faster and providing strong, predictable rewards to companies that invest billions of dollars into medical discoveries. The continent is modernizing the evaluation of medicines throughout the entire life cycle of a drug.
A major feature of this update is its handling of regulatory data protection. When a pharmaceutical company invents a brand-new medicine, they receive a specific window of time during which competitors cannot copy their laboratory data to make cheap, generic versions. The new framework modernizes this timeline by offering extended data and market protection terms under specific conditions.
If a company creates a drug that treats a totally unaddressed disease, or if they launch the drug across every single European member state simultaneously, they can earn extra months of exclusive market control. This clever setup uses market access as a reward, encouraging companies to launch their newest treatments in smaller or less wealthy nations instead of just focusing on the biggest, richest markets.To explore the official steps and structural timelines of this historic legal rewrite, visit the European Medicines Agency Legislation Reform Portal.
Managing Supply Obligations and Preventing Shortages
Another major pillar of the new European rules targets a problem that has plagued hospitals for years: medicine shortages. We have all seen news stories about pharmacies running out of basic antibiotics or critical cancer therapies. The updated European rules address this by creating strict, legally binding rules for managing supply obligations.
Pharmaceutical companies are now required to monitor their supply chains with extreme precision. If a factory faces a production delay, the company must alert health authorities months in advance. They must also maintain emergency reserve stocks of critical medicines.
While these rules protect public health, they add a heavy layer of administrative work for drug developers. Companies can no longer just focus on science; they must build highly advanced data-tracking systems to prove to European regulators that their supply chains are secure and resilient.
Local Divergence: The Secret Layer of Review Delays
Even though the master rules are becoming unified across the European continent, real-world drug development often trips over a quieter, localized hurdle. A major central agency like the European Medicines Agency might give a new drug a green light, but a company cannot automatically hand that medicine to a patient.
Before any clinical trial can begin or any new therapy can be integrated into a local hospital, the project must pass through country-specific ethics committees. These local boards review studies to ensure human participants are treated fairly and that local cultural and legal boundaries are respected.

This is where true divergence shows its face. An ethics board in Germany might have a completely different requirement for patient data privacy compared to an ethics committee in Spain or Italy. One country might demand that patient consent forms be written in a highly specific linguistic style, while another might require extra blood tracking tests that the original master protocol did not include.
These local variations create a hidden labyrinth of review layers. If a pharmaceutical firm does not anticipate these regional differences, they will face sudden, cascading delays that can stall a global drug launch for months.
Navigating Local Intelligence and Change Management
Because local rules can vary so wildly, pharmaceutical companies are shifting how they plan their global operations. They are relying heavily on early local regulatory intelligence. This means that years before a drug is even ready for final approval, experts are on the ground in individual countries tracking the specific, unwritten preferences and shifting habits of regional boards.
To see how these international compliance standards are managed digitally across different regions, you can check the systems designed by PSC Software Compliance Systems. This type of specialized data tracking helps companies log changing regional requirements in real time, ensuring that an update made to a study protocol in one nation does not accidentally break a compliance rule in another.
Having local intelligence is only half the battle. A company must also possess effective change management systems. When an ethics committee in one country demands a sudden modification to a clinical trial or a manufacturing process, that change can trigger a massive chain reaction.
The company must update its laboratory records, notify packaging plants, alter digital labels, and inform clinical investigators worldwide. Without a rock solid change management strategy, a single localized request can cause a total operational breakdown across the entire global pipeline.
The Landscape of Drug Discovery Trends
As we advance through 2026, these regulatory shifts are fundamentally altering modern drug discovery trends. In the past, scientists would discover a molecule, test it in an isolated lab, and hand it off to a legal team to deal with compliance later. That old, disconnected model is no longer sustainable.
Today, regulatory strategy is baked directly into the earliest phases of scientific research. If a research team knows that Europe offers massive incentives for treating unmet medical needs, they will deliberately steer their laboratory focus toward rare, untreatable conditions.
Similarly, because supply chain tracking is now a strict legal requirement, discovery teams are choosing chemical ingredients that are easy and reliable to source globally, avoiding rare components that might trigger a supply chain alert later. The legal framework is actively shaping the molecular science of modern medicine.
Harmonization and Divergence in a Connected World
The ultimate goal for global health remains a world where a safe medicine can be developed once and deployed everywhere without unnecessary friction. Steps like the European pharma package prove that large-scale harmonization is possible, turning a collection of independent countries into a predictable, unified marketplace for healthcare innovation.
Yet, the persistence of local ethics boards, regional legal updates, and country-specific demands reminds us that complete uniformity is an illusion. True success in modern pharmaceuticals requires a delicate balance of macro and micro strategies.
Companies must design their master plans to align with sweeping global agreements, while keeping their operational teams agile enough to respect and adapt to local differences. By mastering this complex balancing act, the medical community can ensure that brilliant scientific discoveries do not get trapped in bureaucratic paperwork, but instead find their way into the hands of the patients who need them most.
Don't let the complex labyrinth of localized compliance, supply chain obligations, and shifting ethics board requirements stall your global drug launch. Contact Metis Consulting Services today

Understanding the New ICH-M14 Safety Guidelines
Today, the pharmaceutical industry is moving toward a massive transformation. A historic change occurred when a global organization called the International Council for Harmonization officially adopted a new framework named the ICH M14 guideline. This framework changes the rulebook for drug safety.

In the Guardrail this week: Explore the pharmaceutical industry's massive paradigm shift from isolated clinical trials to real-world data.
By Michael Bronfman
July 6, 2026
Imagine walking into a doctor's office, picking up a prescription, and knowing that the medicine you are about to take is being monitored by a global web of digital information. For decades, the gold standard for testing medicines has been the traditional clinical trial. In those trials, scientists test a new drug on a small, highly selected group of people under perfect conditions. This process works well, but it does not always show how a drug performs in the messy, complicated real world, where people forget to take pills, have multiple health conditions, or mix different prescriptions.
Today, the pharmaceutical industry is moving toward a massive transformation. Medical tracking is shifting from isolated labs to everyday life, powered by real-world data. This data includes everything from electronic health records kept by hospitals to insurance claims and tracking apps. When researchers analyze this everyday data, they generate real-world evidence that provides a clearer picture of how drugs affect diverse populations.
A historic change occurred when the global organization, the International Council for Harmonization, officially adopted a new framework, the ICH M14 guideline. This framework changes the rulebook for drug safety. It elevates everyday medical information into a form of regulatory currency, meaning health authorities now treat this tracking data with the same respect as traditional laboratory research. This shift changes the future of medicine, creating new ways to develop treatments while presenting major challenges regarding data privacy and access.
Understanding the New Safety Guidelines
To understand why this is such a major shift, it helps to look at how medicine tracking used to work across different borders. In the past, if a pharmaceutical company wanted to demonstrate that a drug was safe in both the United States and Europe, it often had to run separate observational studies in each region. Different countries had different rules about what made data reliable, how statistical math should be done, and how reports should be written. This fragmentation slowed down safety checks and made life-saving drugs take longer to reach patients who needed them.
The new global standard solves this problem. This agreement brings the world's major health authorities onto the same page, including the United States Food and Drug Administration and the European Medicines Agency. The official policy is detailed directly at ich.org, which explains how countries are unifying their rules. By creating a single set of expectations, a study built in one country can now be accepted by regulators worldwide.
This framework specifically targets non-interventional studies. These are research projects where scientists do not give patients a new drug or alter their treatment. Instead, researchers simply look backward or watch from a distance, studying how a medicine behaves as people use it naturally. Because these studies rely on information that already exists in hospital databases or pharmacy logs, having a strict global standard ensures nobody cuts corners or manipulates the findings.
Why Pre-Specification is the Key to Trust
One of the biggest concerns with observational research is a practice known as data dredging or cherry picking. Imagine a researcher looking through millions of patient records without a clear plan. If they look long enough, they might find a random pattern that makes a drug look incredibly safe or dangerously harmful, even if that pattern is just a coincidence.
The new framework eliminates this risk by mandating protocol pre-specification. This means that before scientists even look at the patient data, they must write down an exact plan detailing what they are looking for, how they will define a side effect, and how they will handle their math. This plan is locked in place so researchers cannot change their questions halfway through the study to get the results they want.
This approach builds public trust and satisfies strict regulators. When pharmaceutical companies submit their findings, they must prove they followed their blueprint perfectly. This level of planning turns casual healthcare records into high-quality scientific proof that can justify keeping a drug on the market or expanding its use to new groups of patients, such as children or elderly populations who are often left out of original clinical trials.

The Elements of Modern Evidence Packages
As these strict standards take hold, the way pharmaceutical companies present their discoveries is changing. The industry is moving away from simple stacks of paper toward dynamic evidence packages. These modern files combine multiple streams of information into a single master profile for a medicine.
A modern evidence package brings together three main components:
Clinical Trial Data: The traditional, highly controlled laboratory tests that prove a drug can work under ideal circumstances.
Real World Evidence: The continuous tracking of millions of patients using the medication in everyday situations to see how it performs across different ethnicities, ages, and lifestyles.
Digital Biomarkers: Measurable data collected from smartwatches, continuous glucose monitors, and wearable fitness trackers that show how a patient responds to a drug hour by hour in real time.
When these three streams merge, regulators get a rich picture of a drug's true impact. For example, a heart medication might show perfect numbers in a traditional lab trial. However, the wearable smart sensors might show that patients feel dizzy for an hour right after taking it, while hospital records might show fewer long term heart attacks. This complete view helps doctors make better decisions and helps pharmaceutical companies spot risks or secondary benefits much faster than before.
The Barriers of Real World Information
While this data-rich future sounds amazing, it faces significant real-world roadblocks. The first major hurdle is that most healthcare data was never designed for scientific research. When a doctor types notes into an electronic medical record or a hospital submits an insurance claim, their primary goal is to treat the patient and get paid, not to run a clinical study.
This reality creates massive problems with missing or messy data. A doctor might forget to record how much a patient smokes, or a hospital might change the way they code a specific disease mid-year. If researchers try to run high-level statistical analyses on broken information, they will get inaccurate results. Turning raw hospital paperwork into fit-for-use data requires an immense amount of cleaning, sorting, and verifying, which takes time and expensive technology.
The second massive obstacle involves access restrictions and data silos. Medical information is highly personal, and laws like the Health Insurance Portability and Accountability Act in the United States protect patient confidentiality. Because of these vital privacy laws, hospital systems, insurance firms, and tech giants often keep their data locked tightly inside their own networks.
Breaking down these walls without compromising patient privacy is incredibly difficult. If a pharmaceutical company cannot access a wide enough pool of data, their study will not represent the whole population. This leaves them unable to meet the strict global standards required by modern regulators.
The Role of Pharmacoepidemiology in Public Health
The science driving this entire movement is pharmacoepidemiology, the study of the uses and effects of drugs in large populations. This field acts as an early warning system for public health. When a new medicine hits the market, it might have been tested on only a few thousand individuals. If a dangerous side effect occurs in only one out of every fifty thousand people, a traditional clinical trial will likely miss it entirely.
Through large-scale tracking, scientists can monitor millions of prescriptions simultaneously. If a sudden spike in kidney issues appears among patients taking a specific arthritis medication, researchers can spot the trend within weeks instead of years. The new standard gives these scientists a clearer roadmap for designing these studies, ensuring their alerts are based on rigorous math rather than false alarms.
For an in-depth look at how these safety networks operate, the European Network of Centers for Pharmacoepidemiology and Pharmacovigilance provides resources showing how global networks cooperate to trace medicine safety across whole continents. This coordinated surveillance saves lives by ensuring that when a drug risk is discovered anywhere in the world, safety warnings are updated immediately everywhere.
How Health Authorities are Implementing the Standard
As we move through 2026, nations are actively weaving this framework into their daily operations. The transition requires regulatory agencies to rewrite their local playbooks to support the shared global model.
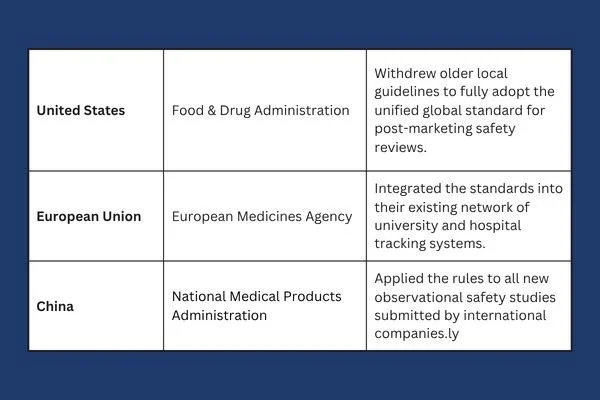
This level of cooperation is rare in international trade, but it shows how vital real world tracking has become. To explore the exact implementation details and view the official updates for American medicine, you can read the documentation here. This page shows how older local frameworks are being replaced to make room for this new way of reviewing drug safety.
The Future of Drug Discovery Trends
Looking ahead, this standard will alter more than just post market safety tracking; it will transform how drugs are discovered and developed from the very beginning. Historically, bringing a single drug to market has taken over a decade and cost billions of dollars. Much of that time was spent waiting for traditional trial results.
By using everyday tracking data as a recognized regulatory asset, companies can now design smarter trials. Scientists can study existing patient databases to discover which specific genetic groups respond best to an experimental treatment before they ever recruit a human volunteer. This approach reduces trial sizes, cuts costs, and protects human participants from taking experimental therapies that are unlikely to help their specific condition.
Furthermore, this framework opens the door for adaptive trials. In these modern studies, researchers can modify an ongoing trial based on incoming real world information, adding new patient groups or adjusting dosages safely with the blessing of regulators. The boundary between the research lab and the everyday clinic is fading away, creating a continuous loop of medical learning.
Balancing Innovation with Ethical Protection
As data becomes the lifeblood of modern medicine, the pharmaceutical industry must handle its new power with caution. Collecting digital footprints from hospital visits, insurance bills, and wrist sensors requires a steadfast commitment to patient ethics. People must be certain that their personal health struggles will never be sold, leaked, or used against them by employers or commercial firms.
The new global standard addresses this by demanding high levels of data transparency and strict data management rules. Companies must explicitly detail how they protect patient identities, strip out personal tracking markers, and secure their databases from cyber threats. Innovation is only valuable if patients feel secure using the systems that monitor them.
The transition to treating real-world information as an official currency marks a massive step forward for human health. It acknowledges that clinical trials, while vital, are just the opening chapter of a drug's true story. By turning everyday experiences into reliable science, the global medical community ensures that the medicines of tomorrow will be safer, more effective, and customized for the real world we all live in.
If your organization needs to convert messy healthcare data into high-quality, audit-ready scientific evidence that meets major global health authorities' requirements, we can help. Contact Metis Consulting Services today