

Outsourcing Oversight: FDA Form 483
Learn why maintaining rigorous third-party quality management is just as vital to your drug pipeline as the science itself. Read about why the critical operational risks highlighted in the FDA Form 483 Warning Letter are important to be aware of, and what dangerous oversight gaps exist in pharmaceutical outsourcing.

By Michael Bronfman
July 20, 2026
This week, discover why maintaining rigorous third-party quality management is just as vital to your drug pipeline as the science itself. Unpack with the "Guard Rail" the critical operational risks highlighted by FDA Form 483 citations and the dangerous oversight gaps in pharmaceutical outsourcing.
Imagine you are running a business that makes life-saving medicines. Because your business is growing fast, you decide to hire outside partners to manufacture the raw chemical ingredients, print the medical labels, and package the final pills. You sign the contracts, assuming these expert partners will handle everything perfectly.
Months later, an official investigator walks into your office and hands you a strict warning paper. The investigator found that your outside partner was using dirty equipment, and because you are the brand owner, your company is legally responsible.
This nightmare scenario happens constantly in the biotech world. The warning paper is called an FDA Form 483, which is the official document issued by investigators when they notice serious violations during factory visits.
A major blind spot in medicine manufacturing involves supplier quality management. Many biotech brands fail to realize that outsourcing production does not mean outsourcing legal liability. When a company fails to watch its external partners closely, it creates an oversight gap.
This gap frequently results in regulatory penalties, frozen pipelines, and missing medicines. Managing supplier risk is no longer just a minor paperwork requirement handled by a background department; it is a critical strategy needed to keep a pharmaceutical pipeline alive.
What is an FDA Form 483 and Why Does It Matter
To understand how this oversight gap happens, you have to understand how government monitoring works. The United States Food and Drug Administration conducts routine, unannounced physical inspections of factories where medicines are created. At the end of an inspection, if the investigator finds problems, they issue a Form 483 list of observations.
A Form 483 is not a final legal punishment, but it is a serious warning. If a business fails to fix the listed problems quickly, the situation can escalate into an official Warning Letter, which can shut down production entirely. You can read about the exact rules for these notices directly on the Inspection Observations Page of the FDA Website. This resource explains how field investigators evaluate factory conditions to ensure public safety.
In recent years, an increasing number of these forms point directly to poor supplier oversight. Government investigators are realizing that while biotech brands claim their products are pure, the brands rarely visit the third-party providers who supply the primary chemical components. This lack of visibility triggers immediate red flags during safety audits.
The Danger of Inadequate Supplier Qualification
The first place the oversight gap appears is during the initial hiring process, known as supplier qualification. Before a pharmaceutical firm buys a single gram of raw material from a third-party provider, they are required by law to run a deep background check on that supplier. This check ensures the provider follows current good manufacturing practices, which are the official quality rules for medicine creation.

Too often, growing biotech firms take shortcuts during this stage. Instead of sending a trained inspector to physically walk through a provider's facility, a firm might simply send a basic text questionnaire through email. They ask the provider if their facilities are clean, the provider checks a box saying yes, and the firm grants them approved status.
This careless approach is called paper qualification, and it fails to satisfy modern regulators. A paper survey cannot spot broken air ventilation systems, leaking pipes, or untrained factory workers. When government inspectors find out that a pharmaceutical company approved a critical material provider based solely on an emailed questionnaire, they issue a severe citation on a Form 483.
Insufficient Oversight of Contract Manufacturers
As biotech companies discover complex new therapies, they rely heavily on specialized partners called Contract Development and Manufacturing Organizations. These partners act as hired factories, taking a drug design from a small startup and mass-producing it inside large industrial facilities.
While outsourcing production makes financial sense, it frequently causes a breakdown in quality control. The biotech brand often adopts a hands-off attitude, believing the hired manufacturer is entirely responsible for daily safety. This is a massive mistake. Under international health frameworks, the brand owner always retains ultimate responsibility for the purity of the drug.
The official global expectations for this relationship are outlined in detail within the FDA Guidance Document on the Q10 Pharmaceutical Quality System. This international framework states that a comprehensive quality management system must extend to the control and review of outsourced activities.
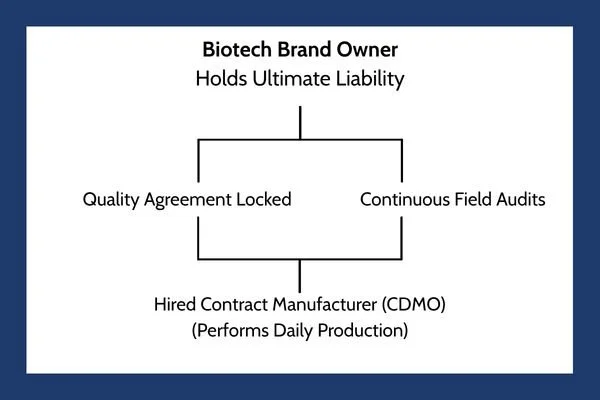
To eliminate the oversight gap, a biotech firm must treat their hired manufacturer as an extension of their own company. This integration requires:
Clear Quality Agreements: A legally binding document that states exactly which company handles specific testing steps, who approves batches, and how mistakes are corrected.
Continuous Person-in-Plant Oversight: Sending a full-time quality expert from the biotech brand to live at the hired manufacturer's facility, monitoring the production lines in person as your drug is made.
Shared Quality Data Tracking: Utilizing unified digital networks so the brand owner can review batch records, laboratory errors, and clean room data in real time, rather than waiting weeks for a text summary report.
The Hidden Risk of Incomplete Supplier Change Notifications
The third area where the oversight gap creates havoc involves supplier change notification processes. Medicine manufacturing is an incredibly delicate science. A microscopic shift in the raw materials can change how a drug behaves inside the human body, altering how fast a pill dissolves or causing unexpected allergic reactions.
Because the science is so sensitive, third-party providers are legally required to inform the biotech brand before making any modifications to their own facilities, raw materials, or testing tools. If a chemical supplier switches to a new raw material provider, or if a label printer updates their machine software, they must submit a formal change notification to the pharmaceutical brand owner.
Unfortunately, communication channels across these supply networks are often broken. A raw material provider might update a chemical purification tool, thinking the change is too minor to mention. If the pharmaceutical brand does not have a strict, mandatory system for tracking and forcing these updates, they remain completely unaware of the alteration.
When a government inspector reviews a biotech company's records and discovers that a raw chemical ingredient was altered without a formal evaluation, the factory receives an immediate Form 483 observation. The brand owner must prove they evaluated every single modification to ensure it did not alter the safety, identity, strength, quality, or purity of the distributed medicine.
Why Quality Management is a Major Pipeline Risk
When a pharmaceutical brand treats supplier oversight as an annoying checklist item rather than a core survival skill, they put their entire business pipeline at risk. The financial and operational damage caused by a single bad supplier can ruin years of scientific research.
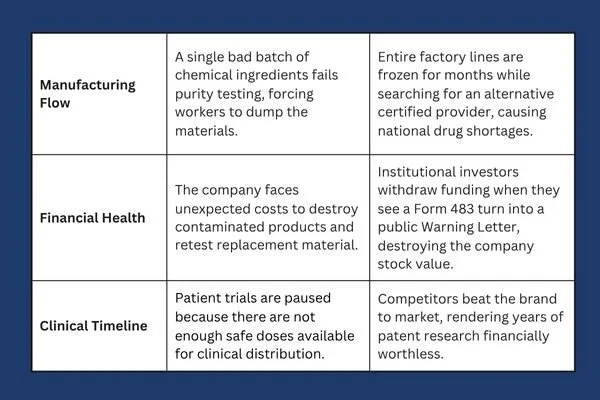
This table shows why supply chain monitoring is a critical commercial necessity. If your external partner fails a government inspection, your entire product launch can stall out, allowing rival businesses to capture the market.
Building a Modern Vendor Review Program
Fixing the oversight gap requires a complete shift in corporate mindset. Biotech organizations must stop choosing their partners based solely on the lowest price or the fastest timeline. Instead, they must construct an aggressive, data-driven vendor review program that treats safety as an active investment.
A modern vendor review strategy replaces casual trust with continuous verification. This process starts by ranking every partner based on risk. A vendor supplying the active chemical ingredient for an injection receives the highest risk rating and requires constant in-person monitoring and quarterly laboratory audits. A vendor supplying external cardboard shipping boxes receives a lower risk ranking, requiring fewer document checks.
Furthermore, companies must establish clear corrective and preventive action programs with their partners. If an external manufacturer makes a mistake, the biotech brand cannot simply accept a brief apology. The brand must force the partner to run a deep root cause analysis to discover why the error happened and prove that systemic changes were implemented to prevent the mistake from ever happening again.
Balancing Rapid Innovation with Supply Security
The biotech world is moving at a breathtaking pace, creating gene therapies and personalized medicines that were science fiction a decade ago. To bring these innovations to reality, companies must rely on a vast web of global suppliers. This interconnected network brings incredible strength, but it introduces immense vulnerability.
The persistent stream of Form 483 citations issued by health authorities is a clear warning that the industry is moving too fast for its own safety infrastructure. Designing an incredible molecule is meaningless if the third-party partner you hire to build it contaminates the batch with residue from a dirty machine.
As we look through 2026, the businesses that survive will be the ones that bridge the oversight gap completely. By treating supplier qualification, active partner communication, and strict change controls as vital priorities, biotech innovators protect their corporate reputation, satisfy tough regulators, and ensure a steady flow of safe therapies reaches the patients who depend on them.
Don't let your outsourcing strategy put your entire pipeline at risk: an unexpected FDA Form 483 or a partner's compliance oversight can derail years of clinical breakthroughs and market value. Contact Metis Consulting Services and establish an ironclad vendor review program, implement excellent quality agreements, and secure your global supply chain against regulatory friction.
Continuous Manufacturing and ICH Q13: Regulatory Readiness at Scale
Guided by the landmark ICH Q13 guidelines, the global pharmaceutical industry is undergoing a revolutionary shift from traditional batch manufacturing to agile, continuous production systems. Read this Week's Guard Rail to explore the revolutionary shift from traditional batch manufacturing to agile, continuous production systems.
Guided by the landmark ICH Q13 guidelines, the global pharmaceutical industry is undergoing a revolutionary shift from traditional batch manufacturing to agile, continuous production systems. Read this Week's Guard Rail to explore the revolutionary shift from traditional batch manufacturing to agile, continuous production systems.
By Michael Bronfman
June 8, 2026
The global pharmaceutical industry is undergoing a major shift in how medicines are made. For many decades, pharmaceutical factories have relied on traditional batch manufacturing. In a batch system, medicine is made in separate steps. Workers mix ingredients in a large tank, stop the machine, transfer the mixture to another station, test it for safety, and then proceed to the next step. This process takes a long time because the materials sit in wait between each phase.
Today, a newer and faster method called continuous manufacturing is changing the field. Instead of stopping and starting, continuous manufacturing moves raw materials through a single, non-stop automated system. Ingredients enter at one end of the factory pipeline, and finished tablets or liquids emerge at the other end.
This modern method affords enormous benefits, but it also creates fresh challenges for global regulators who must ensure every pill is completely safe. To help factories adopt this technology, international experts developed a set of specific rules known as the ICH Q13 guidelines. This system is helping factories around the world upgrade their machinery while keeping patient safety as the top priority.
What is Continuous Manufacturing?
To understand this industrial evolution, it helps to think about how a modern car factory works. Cars are not built by hand one at a time in separate rooms. Instead, they move down an assembly line where any part is added in a continuous, smooth flow. Continuous manufacturing applies this exact same logic to chemistry and medicine.
In a traditional batch setup, if a company wants to produce 1 million doses of a drug, it might need to run 5 separate batches. Each batch requires its own setup, cleaning schedule, and quality testing. If something goes wrong during step three of batch two, the entire batch may have to be scrapped, costing the company time and money.
Continuous manufacturing eliminates those separate steps. Machines run constantly for days or weeks at a time. Raw chemical powders are fed into the system at a precise rate and blended by automated mixers. Then they are compressed into pills and continuously coated.
This constant flow improves production. It also requires a much smaller factory footprint. A continuous manufacturing facility can often fit into a room one-third the size of a traditional batch factory, reducing energy use and building costs.
The Challenge of Process Validation and Lifecycle Management
Because continuous manufacturing runs dynamically, it cannot be monitored using old methods. In a batch system, a scientist can walk up to a large tank, scoop out a sample of powder, and take it to a lab to test its purity. In a continuous system, the material is constantly moving through pipes and tubes at high speeds. Stopping the machine to take a sample would ruin the entire production run.
This active flow elicits crucial questions concerning process validation. Process validation is the collection of data that proves a manufacturing process can reliably produce safe, high-quality medicine. Regulators require pharmaceutical companies to prove that their systems are always under control.
To achieve this control, factories use advanced tools known as Process Analytical Technology. Instead of taking physical samples, engineers place optical sensors directly inside the production pipes. These sensors use infrared light and lasers to inspect the chemical makeup of the moving powder in real time.
If the mixture deviates even slightly from the correct formula, the computer system detects the error instantly. The system can then automatically adjust the feeders' speeds or divert the flawed material to a waste bin without stopping the rest of the production line.
Managing this technology over time is known as lifecycle management. As machines age, sensors can lose accuracy, and software needs to be updated. Pharmaceutical companies must have strict plans in place to maintain, test, and calibrate these digital instruments throughout the entire lifespan of the manufacturing line.
Understanding ICH Q13 and Global Regulatory Harmony
Because different countries have their own individual health ministries, pharmaceutical companies routinely face a confusing web of rules. A factory design that is approved in the United States might face different questions from regulators in Europe or Japan. This lack of agreement can delay the release of important global medicines.
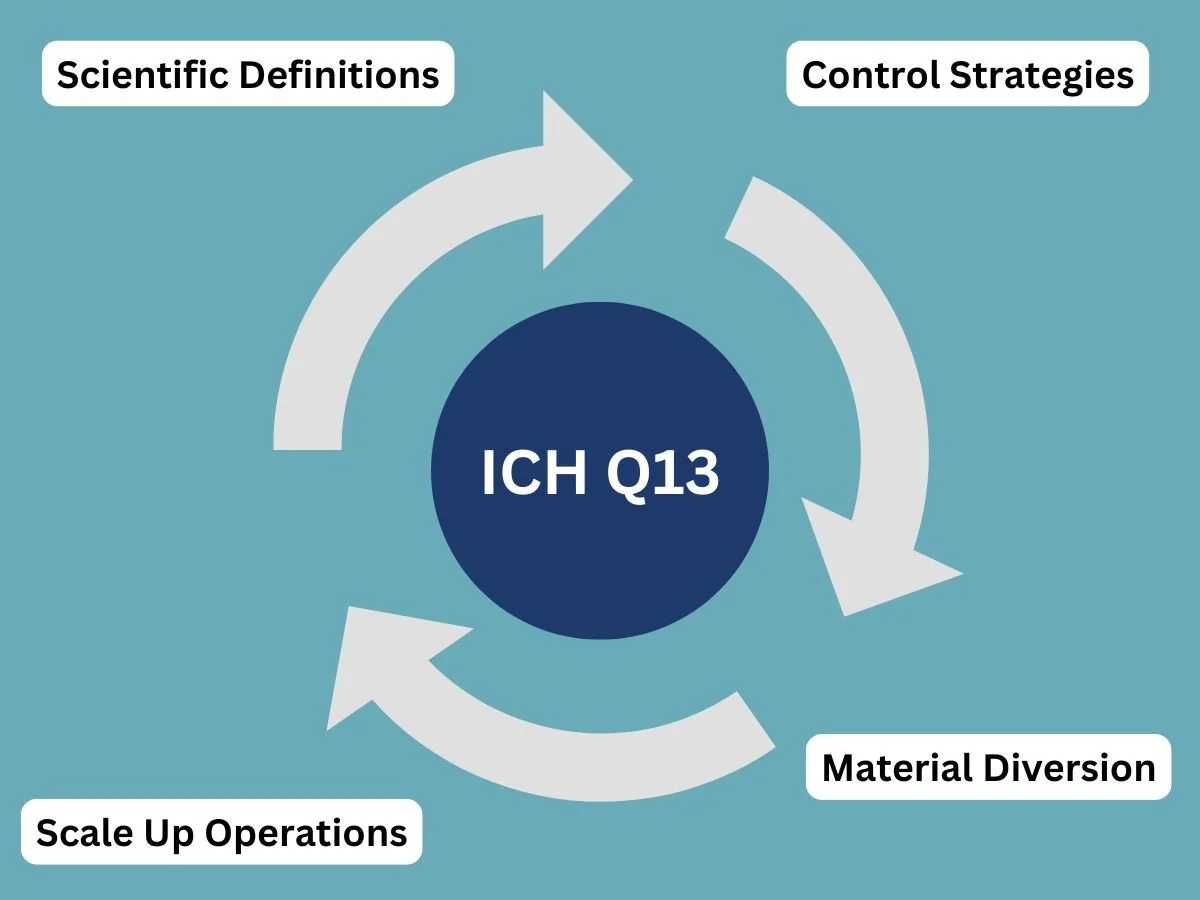
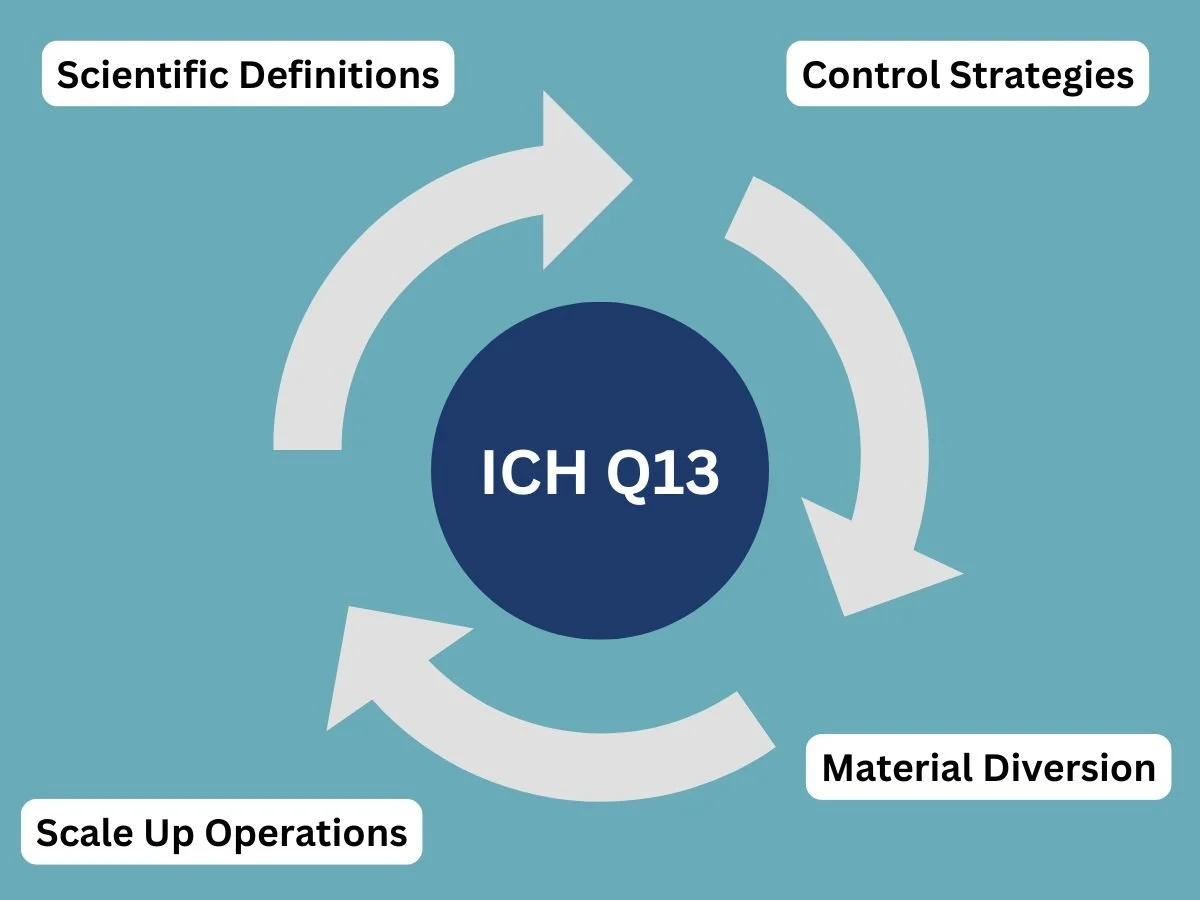
To solve this issue, the International Council for Harmonization created the ICH Q13 guideline. The goal of this document is to establish a single, internationally accepted standard for continuous manufacturing. You can read the specific technical details and formal announcements by visiting the ICH Guidance Documents page.
The ICH Q13 framework gives unambiguous instructions on how companies should handle key manufacturing concepts, including:
Scientific Definitions: Defining exactly what constitutes a batch when the material never stops flowing.
Control Strategies: Explaining how to use real-time sensors to monitor product quality.
Material Diversion: Setting rules for how and when a machine should discard substandard materials during production.
Scale Up Operations: Explaining how a company can increase production volume by simply running the machines longer, rather than building larger equipment.
By setting up these uniform rules, ICH Q13 brings global regulatory readiness to scale. It provides health inspectors with a clear checklist for reviewing these advanced facilities, thereby speeding up and making the approval process more predictable for everyone involved.
Helping Smaller Pharmaceutical Companies Innovate
In the past, only the largest global pharmaceutical corporations had the money and scientific expertise to build continuous manufacturing lines. These projects required millions of dollars in custom engineering and hundreds of hours of consultation with regulatory experts to demonstrate that the systems were safe.
The arrival of the ICH Q13 guidelines changes the landscape. Because the rules are now clearly written down and agreed upon by global authorities, the path to implementation is much easier to follow. This foreseeability makes it feasible for smaller pharmaceutical companies with less internal expertise to employ this manufacturing approach.
Instead of designing a system from scratch, smaller manufacturers can purchase pre-validated equipment that already meets international standards. They can look at the ICH Q13 document as a step-by-step blueprint for compliance. This opening of technology means that smaller companies specializing in rare diseases or generic medicines can also benefit from the efficiency, speed, and cost savings of continuous production.
Enhancing Drug Supply Chain Resilience
One of the greatest benefits of shifting to nonstop production is its contribution to the global drug supply chain. The medical world frequently faces drug shortages caused by factory delays, contaminated batches, or sudden spikes in demand during public health emergencies.
Traditional batch manufacturing is slow to react to these crises. If a hospital suddenly needs double the amount of a specific antibiotic, a batch factory has to source more raw ingredients, schedule new production slots, and run multiple separate batches over several weeks.
Continuous manufacturing solves this problem through flexibility. To scale up production in a continuous facility, you do not need to buy bigger tanks or redesign the process. You simply keep the existing machines running longer. If a machine is scheduled to run for twenty-four hours, engineers can keep it running for seventy-two hours instead.
This ability to rapidly scale production helps prevent shortages and assures that life-saving medicines remain available to patients during emergencies. For perspectives on how these supply chain improvements are being integrated into the wider medical field, you can review current industry analysis on the ISPE Continuous Manufacturing Resources Portal.
The Future of Pharmaceutical Engineering
As more factories adopt continuous manufacturing and follow ICH Q13 standards, the entire pharmaceutical domain will continue to evolve. We are already seeing the integration of fabricated intelligence along with machine learning into these automated lines. Computers can now analyze data from thousands of sensors simultaneously, predicting when a mechanical part might fail before it actually breaks down.
This high level of automation also reduces human error. Because humans do not need to manually scoop powders or transfer materials between stations, the risk of accidental contamination drops drastically. The entire process becomes cleaner, safer, and more efficient.
The transition from batch production to continuous manufacturing represents a true revolution in pharmaceutical engineering. While adjusting to these flexible validation tools entails considerable effort from both scientists plus regulators, the rewards are clear. Through international cooperation and guidelines such as ICH Q13, the pharmaceutical industry is building a more durable, scalable, and reliable system for protecting human health worldwide.
To better understand how this digital evolution affects the greater healthcare sector, we must examine how regulatory readiness shapes the commercial market. When factories adopt advanced automated systems, they do not just change their internal mechanics. They alter how quickly new therapies can reach the market.
For a closer look at how these manufacturing advancements affect actual product availability and commercial rollouts, you can track the latest pharmacy inventory updates. This connection shows that factory-floor innovation directly affects what is available on local pharmacy shelves.
Training the Next Generation of Specialists
As the industry transforms away from manual methods, the training required for pharmaceutical workers is also evolving. The modern factory floor looks more like a high-tech computer lab than a traditional chemical mixing plant.
Engineers must be fluent in data assessment, software maintenance, and mechanical engineering. They need to understand how to read complex up-to-the-minute data streams to spot microscopic variations in product density or moisture levels.
This demand for highly specialized skills has led to new partnerships between universities and industrial leaders. Educational programs are updating their chemistry and engineering courses to focus heavily on continuous processes and international regulatory frameworks.
By training students on the exact tools used in modern automated facilities, the academic world ensures that the workforce is fully prepared to operate complex systems. This educational pivot helps smaller businesses build internal expertise without hiring expensive outside consulting firms.
A Cleaner Blueprint for Global Health
Finally, the combination of advanced technology and clear international rules provides a cleaner, progressively sustainable blueprint for global public health. By limiting waste, reducing factory energy requirements, and dropping the rate of failed batches to near zero, continuous production creates a much more reliable pharmaceutical infrastructure.
When a factory runs smoothly without interruptions, manufacturing costs drop, ultimately assisting the individual patient paying for prescriptions.
The ongoing harmonization of these rules means that a breakthrough discovered in one corner of the world can be rapidly scaled up and manufactured across multiple continents using the exact same validated guidelines. This level of global readiness ensures that humanity is better prepared to address future health challenges quickly, efficiently, and in accordance with strict safety standards.
Ready to seamlessly transition your company through the complexities of ICH Q13 and the process validation, regulatory compliance, and on to the future of pharmaceutical engineering. Contact Metis Consulting Services today.
ICH Q12: Post-Approval Change Management for Pharmaceutical Product Lifecycle Management
The formalized roll-out of ICH Q12 mechanisms introduces an organized approach to identifying and managing regulatory commitments. This framework allows manufacturers to execute routine modifications under the oversight of their internal Pharmaceutical Quality System (PQS), reducing the burden of prior-approval regulatory filings.
This week in the Guardrail, we break down the practical mechanics of the ICH Q12 framework. Tools like Established Conditions and Post-Approval Change Management Procedures can streamline regulatory paths and protect global supply chains.
By Michael Bronfman
May 29, 2026
The global pharmaceutical regulatory framework is transitioning from a rigid, reactive paradigm to an anticipatory, science and risk-based lifecycle management model. Central to this transformation remains the international implementation of the International Council for Harmonization (ICH) Q12 guideline.
Historically, post-approval changes (PACs) to chemistry, manufacturing, and controls (CMC) required extensive, multi-jurisdictional regulatory reviews. These extended processes frequently delayed the introduction of manufacturing innovations, equipment upgrades, and site transfers.
The formalized roll-out of ICH Q12 mechanisms introduces an organized approach to identifying and managing regulatory commitments. This framework allows manufacturers to execute routine modifications under the oversight of their internal Pharmaceutical Quality System (PQS), reducing the burden of prior-approval regulatory filings.
Evolving Jurisdictional Implementation Boundaries
Global regulatory bodies are adopting the tools and enablers outlined in ICH Q12 at varying paces and within specific product domains.
Health Canada Strategy
Health Canada has introduced updates to its regulatory infrastructure, denoting a step-wise integration of the ICH Q12 framework. The Biologic and Radiopharmaceutical Drugs Directorate (BRDD) updated its Health Canada Guidance on Post-Notice of Compliance Changes Framework to establish the operational boundaries for these tools.
Initial implementation focuses exclusively on Post-Approval Change Management Procedures (PACMPs) for products regulated by the BRDD, including biologics and Schedule C drugs. Under this system, the submission of qualifying PACMPs will be formally accepted following a 90-day transition period ending August 13, 2026.
Notably, Established Conditions (ECs) for all product classes and PACMPs for applications outside the BRDD fall outside the initial scope. Broader integration by the Pharmaceutical Drugs Directorate (PDD) is scheduled for subsequent phases, with particular timelines expected later in the year.
Global Agency Status
The United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) provide precedents for these tools across chemical entities and therapeutic biologics. These agencies accept regulatory submissions containing explicitly defined ECs and PACMPs, provided the manufacturer demonstrates an advanced PQS during routine facility inspections.
The differences in implementation speed underscore the need for multinational pharmaceutical operations to design global change strategies that navigate different regulatory requirements.
Functional Mechanics of ICH Q12 Regulatory Tools
The practical value of ICH Q12 relies on two interconnected instruments: Established Conditions and Post-Approval Change Management Guidelines. These tools shift the focus of regulatory dossiers from arbitrary data elements to critical-to-quality variables.
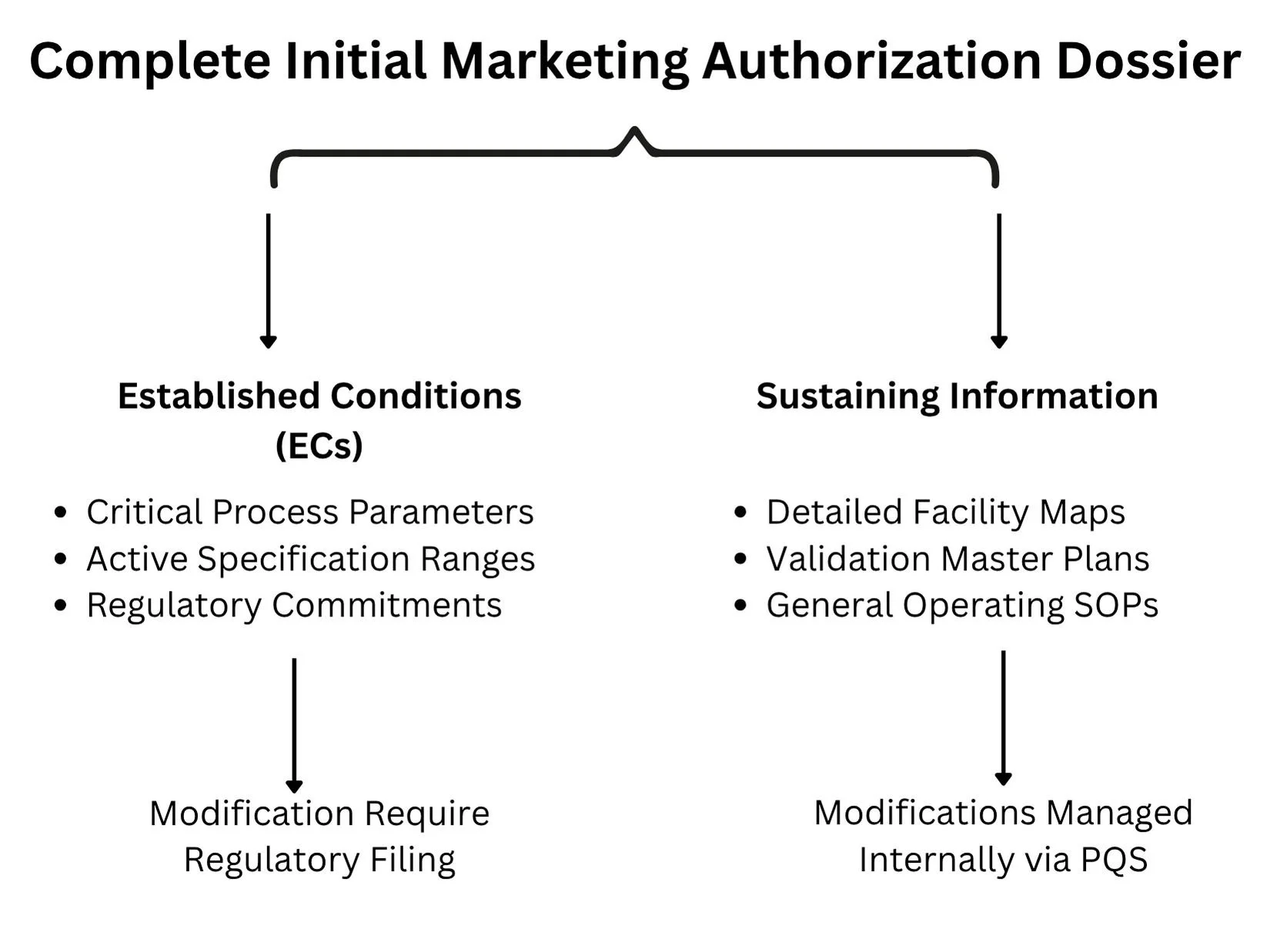
Delineating Established Conditions from Sustaining Information
A longstanding challenge in lifecycle management has been the lack of a clear distinction between legally binding regulatory commitments and purely illustrative descriptive text in the Common Technical Document (CTD). ICH Q12 tackles this by separating information into Established Conditions and Sustaining Information.
Established Conditions (ECs): Defined as the specific elements of a manufacture or control strategy necessary to ensure product safety, identity, strength, purity, or potency. Any modification to an approved EC constitutes a regulatory change that must be reported to the oversight agency. Examples include critical process parameters (CPPs), critical quality attributes (CQAs), acceptance criteria for raw materials, and active operational dimensions of specialized purification columns.
Sustaining Information: Encompasses the underlying science, developmental data, and operational context that supports the designation of ECs. This includes detailed facility blueprints, validation master plans, general operating standard operating procedures (SOPs), and experimental data from early pilot scales. Modifications to sustaining information do not require regulatory notification and are managed entirely via internal site change control protocols.
Post-Approval Change Management Procedures (PACMPs)
A PACMP is a comprehensive, legally binding plan that details a specific manufacturing modification that the sponsor intends to implement throughout the product lifecycle. The protocol explicitly outlines:
The exact nature of the proposed modifications (such as changing an analytical method, upgrading a bioreactor configuration, or transferring an active pharmaceutical ingredient to an alternate facility).
The risk-management strategy is used to evaluate the possible impact of the modification on product quality attributes.
The specific analytical testing, validation matrix, and stability commitments are required to show product comparability.
The predetermined down-regulated reporting category (for example, converting what would typically be a prior-approval Supplement into a post-implementation Notification) if all specified acceptance criteria are met.
By securing prior agency approval for the testing methodology and downgrading logic in the initial PACMP submission, manufacturers can implement modifications quickly once internal testing confirms success.
Structural Requirements of a Mature Pharmaceutical Quality System (PQS)
Sponsors cannot utilize the regulatory flexibilities of ICH Q12 without showing a functional, highly capable PQS that complies with ICH Q10 principles. Regulatory bodies will not grant down-regulated change pathways to facilities lacking robust, data-driven internal quality governance.
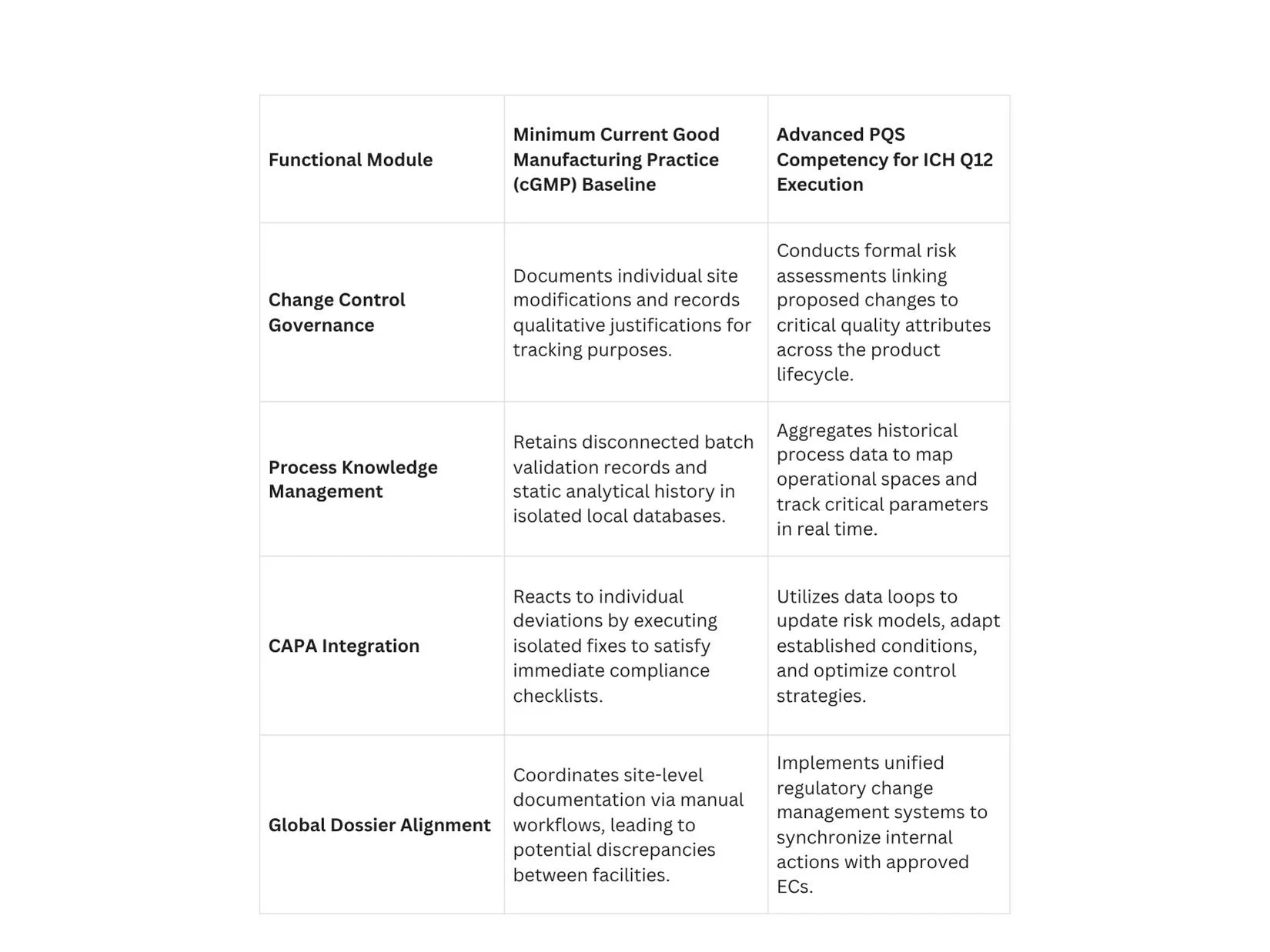
Process Knowledge Management across the Lifecycle
A compliant PQS must operate a continuous knowledge management framework that captures data from early clinical development through commercial manufacturing. Process knowledge should not be stored in isolated paper batch records or disparate local databases.
Instead, it must be aggregated into unified data structures that clearly reflect process parameters, material sources, and environmental variables. This deep process knowledge provides the scientific basis for proposing, justifying, and defending specific boundaries for Established Conditions during regulatory audits.
Regulatory Reporting Categories and Downgrading Strategies
The implementation of ICH Q12 provides a mechanism to modify the default regulatory reporting structures defined by regional laws. The goal is to move low-risk, well-understood adjustments out of prior-approval queues and into post-implementation notification pathways.
The Mechanism of Risk-Based Downgrading
When a manufacturer demonstrates an extensive understanding of a process, they can propose a risk-based categorization strategy for individual ECs. For instance, a process parameter with a broad operating margin and minimal impact on structural attributes can be negotiated from a major reporting tier down to a minor tier.
When this strategy is combined with an approved PACMP, the process efficiency increases significantly. A site transfer for a complex biologic that traditionally required a detailed, prior-approval application can be executed as a post-notice change, provided the verification data satisfies the criteria defined in the protocol.
Introduction of Immediate Notification Pathways
To support such flexibility, modernized regulatory revisions are launching new communication mechanisms. For example, Health Canada introduced a Level III Immediate Notification category within its updated framework. This reporting tier accommodates modifications that have been downgraded from higher risk categories via approved ICH Q12 enablers.
Sponsors utilizing this pathway must notify the agency within 15 days of releasing the modified product to the Canadian market, allowing the regulatory body to maintain oversight without delaying commercial supply lines.
Technical Step-by-Step Implementation Protocol for PACMP Execution
Successfully executing an approved PACMP requires strict adherence to a systematic operational workflow to preserve compliance throughout the product lifecycle.
Phase 1: Protocol Development and Submission
The sponsor prepares a comprehensive PACMP submission within the initial marketing authorization application or via a subsequent formal variation supplement. This document must include a precise description of the future change, the risk mitigation strategy, the analytical methods to be used, and the targeted down-regulated reporting category. The protocol must be reviewed and approved by the target regulatory authority before any subsequent lifecycle modifications can use this pathway.
Phase 2: Internal Facility Execution and Validation
Once the protocol is approved, the manufacturer can initiate the physical change at the designated facility. For example, if transferring production to a new manufacturing line, the site must install the equipment, execute installation and operational qualifications, and run commercial-scale comparison batches.
All analytical data, validation outputs, and stability testing must be conducted exactly as specified in the approved PACMP.
Phase 3: Data Comparison and Acceptance Verification
The quality unit compiles the resulting analytical data and evaluates it against the predetermined acceptance criteria established in the protocol.
If all parameters fall within the approved boundaries, the change is considered successful. If any metric fails to meet the criteria, the protocol becomes invalid for that modification, and the change must revert to the standard prior-approval submission pathway.
Phase 4: Post-Implementation Reporting
Upon verifying compliance, the manufacturer implements the change in commercial production. The sponsor then files the required regulatory notice under the agreed-down-regulated category, such as an immediate notification or an annual report, citing the approved protocol reference number and providing the supporting validation data package.
Commercial and Operational Impact on Global Supply Chains
The transition to an ICH Q12 framework delivers significant strategic and commercial gains that extend beyond basic regulatory compliance.
Mitigating Drug Shortages through Agility
A primary driver of ICH Q12 adoption is the prevention of pharmaceutical supply interruptions and critical drug shortages. In traditional regulatory systems, expanding manufacturing capacity or onboarding an alternative raw-material supplier could take months due to backlogs in prior-approval queues.
By using approved PACMPs and clearly delineated ECs, manufacturers can activate backup manufacturing facilities and alternative material pipelines within days. This nimbleness secures a continuous supply of critical therapeutics to global markets.
Accelerating Ongoing Enhancement and Innovation
The traditional oversight model inadvertently penalized innovation by mandating extensive regulatory filings for minor process improvements. This administrative burden frequently led manufacturers to run outdated processes rather than handle the complex post-approval review landscape.
ICH Q12 removes these barriers, enabling companies to continuously optimize production lines, implement real-time release testing, and deploy advanced process analytical technologies (PAT) under internal PQS controls. This continuous optimization drives lower operating expenses, reduces batch failure rates, and increases overall manufacturing yields.
Conclusion: The Strategic Criticality of Operationalizing ICH Q12
The implementation of ICH Q12 marks a fundamental shift toward an optimized, data-driven approach to pharmaceutical lifecycle management. By implementing tools such as Post-Approval Change Management Procedures and explicitly mapped Established Conditions, manufacturers can significantly reduce regulatory timelines and accelerate facility upgrades.
However, these advanced regulatory flexibilities cannot operate in a vacuum; they require a highly capable, data-driven Pharmaceutical Quality System built on robust knowledge management and risk-based decision-making. As regulatory authorities globally continue to embed these guidelines into their standard oversight frameworks, companies that fail to operationalize these enablers risk a permanent competitive and operational disadvantage.
To review the scientific consensus, emerging clinical data, and peer-reviewed case studies supporting advanced lifecycle management strategies, quality professionals can access Nature.com to ensure their operational systems comply with current international practices.
Don't let rigid regulatory frameworks hold back your manufacturing innovation or compromise your supply chain stability. Contact Metis Consulting Services today.

AI in Regulatory Submissions: Writing for Both Human and Machine Reviewers
This week in the Guardrail, we analyze the dual-audience reality facing modern pharmaceutical compliance. As regulatory agencies integrate automated tools to parse complex submissions, drug sponsors must adapt their documentation strategies to satisfy both algorithmic logic and human expertise.

This week in the Guardrail, we analyze the dual-audience reality facing modern pharmaceutical compliance. As regulatory agencies integrate automated tools to parse complex submissions, drug sponsors must adapt their documentation strategies to satisfy both algorithmic logic and human expertise.
By Michael Bronfman
May 25, 2026
The world of making and approving medicines is going through a massive shift. For decades, pharmaceutical companies wrote drug applications for just one audience: human scientists. Teams of medical doctors, chemists, and statisticians at agencies like the Food and Drug Administration would read thousands of pages of text to decide if a new drug was safe.
Today, that process looks very different. Pharmaceutical companies now use computer algorithms, known as Artificial Intelligence, to run clinical trials and analyze data. At the same time, the regulatory agencies themselves are starting to use computer programs to help read and sort through massive piles of application documents.
This means medical writers and drug sponsors must now write for two very different audiences at the same time. They must write for the human experts who make the final decisions, and they must write for the machine reviewers who scan the text for errors and patterns. If an application is not structured correctly for a machine to read, it could get flagged for inconsistencies before a human expert even looks at it.
To help companies navigate this change, the Food and Drug Administration released official draft guidance about using these advanced computer models in drug development. This document outlines exactly how the agency looks at data generated by computers and how companies should share that information. For more detailed context, you can read the official announcement on the FDA Press Release Page.
The Food and Drug Administration Risk Framework
The official policy from the government makes one thing very clear: not all computer applications are treated equally. The agency uses a risk-based framework to grade how much scrutiny a system needs. This framework is based on two main ideas: model influence and decision consequence.
Model influence means how much the computer output affects the final decision. If a computer makes a final choice on its own, its influence is strong. If a human expert checks the work and can override the computer, its influence is lower. Decision consequence means what could go wrong if the computer makes a mistake. If a computer error harms a patient, the consequences are high. If an error just slows down a factory machine for an hour, the consequence is low.
By looking at these two factors, the government separates computer tools into high-scrutiny systems and low-requirement systems.

High Scrutiny Systems
The highest level of official review is saved for computer systems that directly create evidence for a drug application. These are systems where a mistake could directly hurt a patient or ruin the results of a scientific study.
The government pays closest attention to these five specific areas:
Patient Stratification: Choosing which patients get to be in a clinical trial based on their genetic codes or medical histories.
Dose Optimization: Using mathematical models to calculate exactly how much medicine a patient should take to get better without getting sick from side effects.
Real World Data Analysis: Scanning millions of electronic health records from hospitals to see how a drug performs in everyday life outside of a controlled trial.
Safety Signal Detection: Watching patient data in real time to spot rare and dangerous side effects before they become a widespread public health crisis.
Endpoint Derivation: Using wearable sensors like smartwatches to measure how well a patient is moving or sleeping during a clinical trial.
If a company uses a computer for any of these tasks, it must prove the system is incredibly reliable. They must show how the model was trained, what data it used, and how it avoids bias.
Low-Requirement Systems
On the other side of the coin, some computer uses do not impact patient safety at all. If a company uses a computer tool to format a document, check page numbers, or organize internal administrative tasks, the government does not need to see piles of validation data. These internal operations face proportionally lower requirements because a mistake by the computer will not change the scientific conclusions of the drug trial.
Understanding the Double Audience
Because regulatory agencies are now using advanced software to help manage incoming applications, drug sponsors must realize they are writing for a double audience. The text must satisfy both the human brain and the computer algorithm.
To see how these two audiences read differently, look at this comparison:
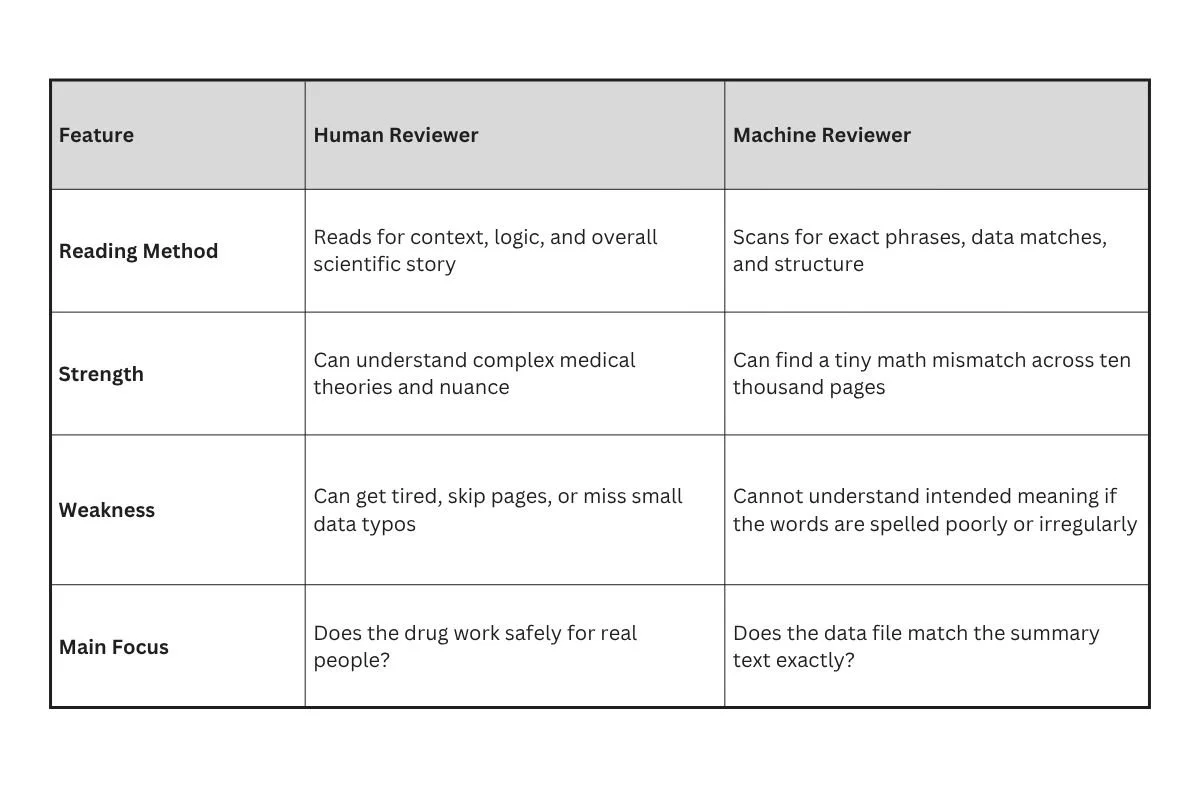
When a human reads a drug application, they want a clear narrative. They want to understand the journey of the drug from the laboratory to the clinic. They care about scientific logic.
A machine reviewer does not care about stories. It treats the document like a database. It looks at the tables, the labels, and the numbers to make sure everything adds up perfectly. If the summary on page five says fifty patients had a headache, but the raw data table on page nine hundred says forty-nine patients had a headache, the machine will flag that instantly. A human might miss that small slip, but a machine never will.
Writing for the Machine Reviewer
Writing for a computer means changing how you present text. Computers like clean organization, predictable patterns, and explicit language. If you write with vague words, the software can get confused and flag your document as a risk.
Structure and Predictability
The best way to help a machine reviewer is to use standard templates. Regulatory documents should follow strict structural rules. Use clear, standardized headings for every section. Do not try to be creative with section titles. If the standard title is Clinical Efficacy, do not change it to How Well the Drug Worked. The computer looks for specific keywords to map the document, and changing those keywords breaks the map.
Data Consistency and Labels
Every data point must look identical throughout the entire file. If you refer to a drug concentration as ten milligrams on one page, do not write it as 10mg on the next page. Choose one format and stick to it.
Also, make sure that every chart and table has clear, descriptive labels that use text instead of scanned images. Machine reviewers read text characters, not picture pixels. If you paste a picture of a table into your document, the computer sees a blank space and misses all the important data inside it.
Front Loading for Clarity
Machines are built to look for core conclusions early. Put your main findings, safety summaries, and essential data points right at the front of your sections. Do not hide your main message under paragraphs of introductory fluff. Front loading your clarity helps the computer categorize your document correctly on its very first pass.
Writing for the Human Reviewer
While you must make your document easy for a computer to analyze, you cannot forget the human being who must sign the final approval paper. Humans need context, clear explanations, and a believable scientific argument.
Explaining the Why
A machine can show that a number changed, but only a human can explain why it changed. If a clinical trial had a sudden drop in patient attendance during month four, a machine might flag it as a data error.
The human writer needs to explain the context:
"Patient attendance dropped in month four due to a historic blizzard that closed three major clinical trial sites for two weeks, but patients resumed their regular visits as soon as the roads cleared."
This explanation satisfies the human reviewer and prevents them from rejecting the data.
Keeping the Story Alive
A good regulatory submission tells a story of safety and success. The human writer must connect the dots between different pieces of research. Show how the animal studies predicted the human results, and show how the human results match the goals of the project. Use active, plain verbs to explain what the scientists did. Avoid overly dense language that puts the reader to sleep. A tired reviewer is a frustrated reviewer.
Conducting an Internal Review
Before you click the submit button to send your drug application to the government, your team should perform a complete internal review. This means testing your document against your own software tools to see what a machine reviewer will find.
Step One: The Automated Consistency Check
Run your completed document through text-matching software. This program should look for every number, percent, and statistical value to make sure they match perfectly across all chapters. If the software finds a conflict, fix it immediately. You want to find these errors yourself rather than letting the government find them first.
Step Two: The Structure Audit
Verify that every hyperlink works and leads to the correct appendix. Check that your document map functions properly and that all headings match the standard table of contents. If a machine cannot navigate your document links, it may automatically reject the file.
Step Three: The Human Readability Pass
Have a scientist who did not write the document read it for flow and clarity. Ask them if the arguments make sense and if the explanations are easy to find. This step ensures that once your document passes the computer gates, it will please the human experts.
The Path Forward for Drug Developers
The use of computer intelligence in regulatory submissions is not a temporary trend. It is the permanent future of medicine. Drug companies that learn how to write for both humans and machines will get their medicines approved much faster. Those who stick to old ways of writing will face constant delays, data flags, and rejection notices.
To keep up with these changes, companies should train their medical writers in basic data science principles. Writers do not need to learn how to code, but they do need to understand how computers read and sort information. By focusing on predictability, exact data matches, and clear summaries, you can create a document that satisfies the cold logic of a machine and the deep wisdom of a human scientist.
To learn more about how the government views these new digital tools, you can review the comprehensive resources provided by theFDA Artificial Intelligence Development Page. Staying informed about these official updates is the best way to ensure your future submissions are successful.

FDA Under DOGE: What Workforce Reductions Mean for the Pharmaceutical Industry
FDA Under DOGE: What Workforce Reductions Mean for the Pharmaceutical Industry

There are seismic shifts occurring within the FDA as DOGE-led workforce reductions redefine the boundaries of regulatory oversight. It is a new era where the burden of pharmaceutical safety is shifting from the government to the private sector.
By Michael Bronfman
May 18, 2026
American healthcare is undergoing a massive shift in 2026. Under the new Department of Government Efficiency (DOGE), the Food and Drug Administration (FDA) has faced a notable transformation. More than 3,500 employees have been let go. These aren't just office workers; they are the scientists, inspectors, and experts who make sure the medicine in your cabinet is safe.
For people working in the pharmaceutical industry, this is a "mission-critical" moment. When the government agency that watches over you loses a large chunk of its workforce, the rules of the game change. You have to understand what a "leaner FDA" means for your daily job and for the patients who count on your products.
The Scale of the Change
To understand the impact, we have to look at who is gone. The cuts have hit almost every part of the agency. We are seeing fewer people in charge of:
Approving drug labels: Making sure the instructions on a bottle are correct and easy to read.
Posting recall notices: Getting the word out quickly when a dangerous product is found.
Testing samples: Actually looking at the chemicals in a lab to verify they match the given recipe.
According to Healthgrades reports, these cuts are already being felt on the ground. When you lose that many people, the wait time for everything starts to grow.
The Ripple Effect on Inspections
In the past, pharmaceutical companies expected regular visits from FDA inspectors. These visits kept everybody on their toes. With a smaller workforce, the FDA cannot be everywhere at once. Currently proposed is a “one-day inspection,” which may not be sufficient time for a regulatory body to carry out a thorough inspection of patient-facing treatment.
Legal experts at Ropes & Gray LLP have noted that workforce reductions will likely lead to longer investigative timelines. If there is a problem at a facility, it might take much longer for the agency to find it, or to clear a company that has resolved an issue. This creates significant uncertainty for sponsor organizations.
The Impact on Global Trade
The FDA doesn't just watch over US manufacturing sites. They also inspect sites globally, including in India and China, that export medicine to the United States.
International Inspections
Travel is expensive and time-consuming. With fewer inspectors, the number of overseas visits has dropped sharply. This creates a risk. If an overseas plant knows it won't be inspected for 5 years, it might get lax about its standards.
Smart companies are now performing their own "Supply Chain Audits." They are sending their own teams to visit their partners worldwide to ensure that every ingredient is pure. You cannot afford to have a partner who cuts corners.
Navigating Internal Reorganizations
The FDA is also being reorganized. Offices are merging, and departments are being renamed. For a pharma company, this means your "point of contact" might change every month.
Tips for Staying Connected
Document Everything: Keep a clear trail of every email and phone call with the agency.
Be Clear and Concise: Since FDA staff are overwhelmed, make your letters easy to read. Use bullet points and put the most important info first.
Monitor the Federal Register: Stay updated on new rules being issued to address the smaller workforce.
The Economic Reality
DOGE’s goal was to save taxpayers' money. While the government is spending less on salaries, the pharma industry might end up spending more.
The industry is learning that "less government" doesn't always mean "less work." It often means the work stops in the approval pathway.
Looking Ahead: The Future of the FDA
The year 2026 will be remembered as a turning point. We are moving toward a model in which the government sets the high-level rules, while companies are expected to police themselves much more strictly.
A New Partnership
The relationship between the FDA and pharma companies used to be like a teacher and a student. The teacher (FDA) would grade the student’s (Pharma) work and tell them how to fix it.
Now, the relationship is more like a judge and a citizen. The judge doesn't have time to teach you. They only have time to show up when something goes wrong and hand out a punishment.
Practical Compliance Steps for 2026
If you want to survive and thrive in this new environment, your team should focus on these three pillars:
1. Data Integrity
Every number in your report must be perfect. Since the FDA will be doing fewer "random checks," they will likely be much harsher when they find a data error. They will assume that if you made one mistake, you are hiding others.
2. Supply Chain Transparency
Know exactly where your Active Pharmaceutical Ingredients (APIs) come from. If your supplier in another country hasn't seen an FDA inspector in three years, you need to be the one to inspect them.
3. Rapid Response Teams
Have a plan ready for when something goes wrong. If you find a safety signal, you need to know exactly how to handle a recall without waiting for the FDA to hold your hand through the process.
The New Mission
The workforce reductions at the FDA are a challenge, but they are also an opportunity. Companies that prove they can maintain high standards without constant government supervision will win the trust of doctors and patients.
For pharmaceutical professionals in quality and regulatory , the mission is the same, but the pressure has increased. You are now the primary protectors of public health. By staying informed through resources like Healthgrades and keeping an eye on legal shifts at sites like Ropes & Gray LLP, you can navigate this leaner landscape more confidently.
The FDA might have fewer people, but the patients still expect the same level of safety. It is up to us to deliver it.
Is your organization ready for a one-day inspection or a supply chain failure? Discover the gaps in your compliance strategy; contact Metis Consulting Services today to fortify your quality systems and navigate the leaner regulatory landscape of 2026 with confidence.
Key Takeaways
The FDA is smaller now. Over 3,500 people lost their jobs, meaning the government has fewer experts to monitor the Pharma field
Wait times are longer. With fewer workers, the FDA may take longer to approve new drugs or investigate safety issues.
Companies have to watch themselves. Since our "government watchdog" is busy, drug companies must hire their own experts to ensure their medicines are safe.
Quality is more important than ever. If a company makes a mistake, it might have to handle cleanup on its own, with little help from the government.
Safety is still the goal. Even with a smaller FDA, making sure patients are safe is the main goal.