

Understanding the New ICH-M14 Safety Guidelines
Today, the pharmaceutical industry is moving toward a massive transformation. A historic change occurred when a global organization called the International Council for Harmonization officially adopted a new framework named the ICH M14 guideline. This framework changes the rulebook for drug safety.

In the Guardrail this week: Explore the pharmaceutical industry's massive paradigm shift from isolated clinical trials to real-world data.
By Michael Bronfman
July 6, 2026
Imagine walking into a doctor's office, picking up a prescription, and knowing that the medicine you are about to take is being monitored by a global web of digital information. For decades, the gold standard for testing medicines has been the traditional clinical trial. In those trials, scientists test a new drug on a small, highly selected group of people under perfect conditions. This process works well, but it does not always show how a drug performs in the messy, complicated real world, where people forget to take pills, have multiple health conditions, or mix different prescriptions.
Today, the pharmaceutical industry is moving toward a massive transformation. Medical tracking is shifting from isolated labs to everyday life, powered by real-world data. This data includes everything from electronic health records kept by hospitals to insurance claims and tracking apps. When researchers analyze this everyday data, they generate real-world evidence that provides a clearer picture of how drugs affect diverse populations.
A historic change occurred when the global organization, the International Council for Harmonization, officially adopted a new framework, the ICH M14 guideline. This framework changes the rulebook for drug safety. It elevates everyday medical information into a form of regulatory currency, meaning health authorities now treat this tracking data with the same respect as traditional laboratory research. This shift changes the future of medicine, creating new ways to develop treatments while presenting major challenges regarding data privacy and access.
Understanding the New Safety Guidelines
To understand why this is such a major shift, it helps to look at how medicine tracking used to work across different borders. In the past, if a pharmaceutical company wanted to demonstrate that a drug was safe in both the United States and Europe, it often had to run separate observational studies in each region. Different countries had different rules about what made data reliable, how statistical math should be done, and how reports should be written. This fragmentation slowed down safety checks and made life-saving drugs take longer to reach patients who needed them.
The new global standard solves this problem. This agreement brings the world's major health authorities onto the same page, including the United States Food and Drug Administration and the European Medicines Agency. The official policy is detailed directly at ich.org, which explains how countries are unifying their rules. By creating a single set of expectations, a study built in one country can now be accepted by regulators worldwide.
This framework specifically targets non-interventional studies. These are research projects where scientists do not give patients a new drug or alter their treatment. Instead, researchers simply look backward or watch from a distance, studying how a medicine behaves as people use it naturally. Because these studies rely on information that already exists in hospital databases or pharmacy logs, having a strict global standard ensures nobody cuts corners or manipulates the findings.
Why Pre-Specification is the Key to Trust
One of the biggest concerns with observational research is a practice known as data dredging or cherry picking. Imagine a researcher looking through millions of patient records without a clear plan. If they look long enough, they might find a random pattern that makes a drug look incredibly safe or dangerously harmful, even if that pattern is just a coincidence.
The new framework eliminates this risk by mandating protocol pre-specification. This means that before scientists even look at the patient data, they must write down an exact plan detailing what they are looking for, how they will define a side effect, and how they will handle their math. This plan is locked in place so researchers cannot change their questions halfway through the study to get the results they want.
This approach builds public trust and satisfies strict regulators. When pharmaceutical companies submit their findings, they must prove they followed their blueprint perfectly. This level of planning turns casual healthcare records into high-quality scientific proof that can justify keeping a drug on the market or expanding its use to new groups of patients, such as children or elderly populations who are often left out of original clinical trials.

The Elements of Modern Evidence Packages
As these strict standards take hold, the way pharmaceutical companies present their discoveries is changing. The industry is moving away from simple stacks of paper toward dynamic evidence packages. These modern files combine multiple streams of information into a single master profile for a medicine.
A modern evidence package brings together three main components:
Clinical Trial Data: The traditional, highly controlled laboratory tests that prove a drug can work under ideal circumstances.
Real World Evidence: The continuous tracking of millions of patients using the medication in everyday situations to see how it performs across different ethnicities, ages, and lifestyles.
Digital Biomarkers: Measurable data collected from smartwatches, continuous glucose monitors, and wearable fitness trackers that show how a patient responds to a drug hour by hour in real time.
When these three streams merge, regulators get a rich picture of a drug's true impact. For example, a heart medication might show perfect numbers in a traditional lab trial. However, the wearable smart sensors might show that patients feel dizzy for an hour right after taking it, while hospital records might show fewer long term heart attacks. This complete view helps doctors make better decisions and helps pharmaceutical companies spot risks or secondary benefits much faster than before.
The Barriers of Real World Information
While this data-rich future sounds amazing, it faces significant real-world roadblocks. The first major hurdle is that most healthcare data was never designed for scientific research. When a doctor types notes into an electronic medical record or a hospital submits an insurance claim, their primary goal is to treat the patient and get paid, not to run a clinical study.
This reality creates massive problems with missing or messy data. A doctor might forget to record how much a patient smokes, or a hospital might change the way they code a specific disease mid-year. If researchers try to run high-level statistical analyses on broken information, they will get inaccurate results. Turning raw hospital paperwork into fit-for-use data requires an immense amount of cleaning, sorting, and verifying, which takes time and expensive technology.
The second massive obstacle involves access restrictions and data silos. Medical information is highly personal, and laws like the Health Insurance Portability and Accountability Act in the United States protect patient confidentiality. Because of these vital privacy laws, hospital systems, insurance firms, and tech giants often keep their data locked tightly inside their own networks.
Breaking down these walls without compromising patient privacy is incredibly difficult. If a pharmaceutical company cannot access a wide enough pool of data, their study will not represent the whole population. This leaves them unable to meet the strict global standards required by modern regulators.
The Role of Pharmacoepidemiology in Public Health
The science driving this entire movement is pharmacoepidemiology, the study of the uses and effects of drugs in large populations. This field acts as an early warning system for public health. When a new medicine hits the market, it might have been tested on only a few thousand individuals. If a dangerous side effect occurs in only one out of every fifty thousand people, a traditional clinical trial will likely miss it entirely.
Through large-scale tracking, scientists can monitor millions of prescriptions simultaneously. If a sudden spike in kidney issues appears among patients taking a specific arthritis medication, researchers can spot the trend within weeks instead of years. The new standard gives these scientists a clearer roadmap for designing these studies, ensuring their alerts are based on rigorous math rather than false alarms.
For an in-depth look at how these safety networks operate, the European Network of Centers for Pharmacoepidemiology and Pharmacovigilance provides resources showing how global networks cooperate to trace medicine safety across whole continents. This coordinated surveillance saves lives by ensuring that when a drug risk is discovered anywhere in the world, safety warnings are updated immediately everywhere.
How Health Authorities are Implementing the Standard
As we move through 2026, nations are actively weaving this framework into their daily operations. The transition requires regulatory agencies to rewrite their local playbooks to support the shared global model.
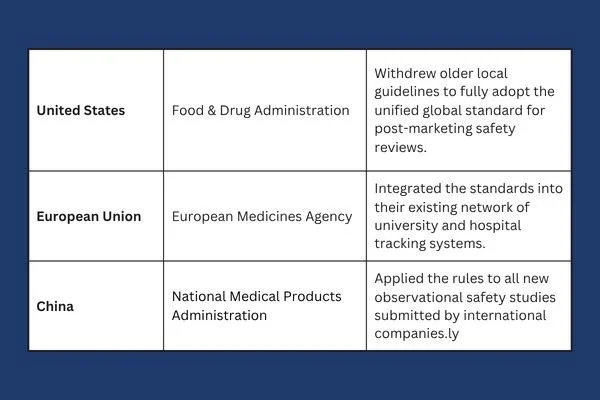
This level of cooperation is rare in international trade, but it shows how vital real world tracking has become. To explore the exact implementation details and view the official updates for American medicine, you can read the documentation here. This page shows how older local frameworks are being replaced to make room for this new way of reviewing drug safety.
The Future of Drug Discovery Trends
Looking ahead, this standard will alter more than just post market safety tracking; it will transform how drugs are discovered and developed from the very beginning. Historically, bringing a single drug to market has taken over a decade and cost billions of dollars. Much of that time was spent waiting for traditional trial results.
By using everyday tracking data as a recognized regulatory asset, companies can now design smarter trials. Scientists can study existing patient databases to discover which specific genetic groups respond best to an experimental treatment before they ever recruit a human volunteer. This approach reduces trial sizes, cuts costs, and protects human participants from taking experimental therapies that are unlikely to help their specific condition.
Furthermore, this framework opens the door for adaptive trials. In these modern studies, researchers can modify an ongoing trial based on incoming real world information, adding new patient groups or adjusting dosages safely with the blessing of regulators. The boundary between the research lab and the everyday clinic is fading away, creating a continuous loop of medical learning.
Balancing Innovation with Ethical Protection
As data becomes the lifeblood of modern medicine, the pharmaceutical industry must handle its new power with caution. Collecting digital footprints from hospital visits, insurance bills, and wrist sensors requires a steadfast commitment to patient ethics. People must be certain that their personal health struggles will never be sold, leaked, or used against them by employers or commercial firms.
The new global standard addresses this by demanding high levels of data transparency and strict data management rules. Companies must explicitly detail how they protect patient identities, strip out personal tracking markers, and secure their databases from cyber threats. Innovation is only valuable if patients feel secure using the systems that monitor them.
The transition to treating real-world information as an official currency marks a massive step forward for human health. It acknowledges that clinical trials, while vital, are just the opening chapter of a drug's true story. By turning everyday experiences into reliable science, the global medical community ensures that the medicines of tomorrow will be safer, more effective, and customized for the real world we all live in.
If your organization needs to convert messy healthcare data into high-quality, audit-ready scientific evidence that meets major global health authorities' requirements, we can help. Contact Metis Consulting Services today
Moving Beyond the Sandbox: How to Build Inspection-Ready AI in Regulated Life Sciences
For years, AI was just a tool for simple tasks like drafting emails. Those days of casual use are over. Regulators are tightening expectations, demanding strict AI governance, perfect traceability, and full integration with quality compliance systems. These rules are no longer optional best practices.
As global regulators rapidly tighten enforcement on pharmaceutical companies, this week we look at the high-stakes gap between flashy AI pilots and the rigorous, audit-ready validation required in the life sciences sector. Manufacturers must have systems in place for bulletproof governance to survive their next inspection.
By Michael Bronfman
June 30, 2026
Many pharmaceutical companies live in a frustrating middle ground. You can see it in boardrooms and IT departments everywhere. Teams enthusiastically say, “We are experimenting with artificial intelligence!” Yet when asked whether that same technology is ready for an official regulatory inspection, the room falls silent.
The gap between a cool pilot project and a fully validated, inspection-ready system is where most life sciences companies currently find themselves.
For years, teams treated artificial intelligence as a collection of non-product tools used for simple tasks. It might have been used to summarize long documents or draft email templates. But the days of casual experimentation are officially over. Regulatory bodies are tightening their expectations. They are demanding strict AI governance, perfect traceability, and complete integration with quality compliance systems. They are no longer leaving these rules to optional best practices.
Artificial intelligence systems inform labeling, product performance claims, drug dosing, patient safety, or quality decisions; they face a tough reality. The entire solution must fulfill rigorous quality, validation, and lifecycle controls. Generic pilots fail to scale because they lack the foundation required to survive a regulatory audit. Life sciences organizations must shift to purpose-built artificial intelligence that incorporates strict governance controls, unalterable audit trails, and validated results to succeed.
The Shift in Regulatory Reality
Why do generic pilots fail?
To understand this, look at how global authorities view technology. The Food and Drug Administration released major updates that signal a strong enforcement posture for advanced software used in regulated spaces. The agency treats high-risk artificial intelligence with the same seriousness as physical medical devices or critical manufacturing tools.
Traditional software validation worked well for static systems. In the past, computer systems validation followed a predictable path. A developer wrote code, a quality team tested that it did exactly what it was supposed to, and the software never changed unless an engineer manually updated it.
Artificial intelligence contradicts this old way of thinking. Advanced models are dynamic. They are built to learn, observe patterns, and develop over time. Because these systems can evolve based on the information they process, standard testing methods are inadequate. Software cannot be tested once and assumed to behave exactly the same way a year from now.
Regulators are fully aware of this challenge. They are looking closely at:
Design Controls: How the model was built, chosen, and structured.
Model Validation: Proof that the mathematical formulas produce accurate, repeatable results.
Data Authenticity: Complete certainty that the information feeding the model is clean and unaltered.
Risk Management: Clear plans for handling unexpected errors.
If an inspector walks into your facility today and sees an advanced tool helping your quality team make decisions, they will ask tough questions. They will want to know how you verify the output. They will want to see how you track changes in the system. If your only answer is that a vendor told you the tool works, you are facing a major compliance risk.
Why Generic AI Pilots Fail to Scale
It is incredibly easy to build a successful pilot project. A small team can upload historical quality data into a popular, generic large language model. Within an afternoon, the tool can review past records and suggest draft standard operating procedures or summarize corrective and preventive action reports. The team celebrates, declares the project a success, and plans to roll it out to the whole company.
Then, they meet the quality assurance department.
Generic artificial intelligence applications are built for mass productivity, not the high-stakes world of life sciences. When you try to push a basic pilot into a Good x Practice environment, the system usually falls apart for several reasons.
The Opaque Decision Process
Generic models operate like a black box. A user submits a question, and the tool provides an answer, but no one can trace the exact path the software took to reach that conclusion. In a regulated environment, an untraceable answer is a non-compliance finding waiting to happen. If you cannot prove how your software reached a conclusion about a batch failure or a clinical trial data point, you cannot use that conclusion.Missing Explicit Intended Use
Validation cannot be generic. You cannot validate an advanced tool for general office work and then use it to triage quality investigations. Every application must have a clearly defined intended use statement. This statement must outline the exact process the tool supports, who the users are, what source systems feed it data, and how the output impacts human health or product quality. Generic tools are not built to restrict themselves to a single, tightly controlled workflow.Model Drift and Information Degradation
When an advanced model interacts with new data, its internal weights can shift. Over time, the system's accuracy can degrade or change, a phenomenon termed as model drift. Generic applications do not include built-in alerts that notify you when the software becomes less accurate. Missing continuous tracking protocols, a tool that worked perfectly during a pilot in January might give flawed recommendations during an inspection in November.Poor Data Lineage and Security
Where does the information go when you type it into a generic tool? Does the vendor use your proprietary molecule data to train their public models? Many basic applications lack clear data lineage. They cannot prove who had access to the data, how it was modified, or where it is stored. This violates fundamental data validity principles that require all records to be fully traceable and secure.
The Core Pillars of True AI Governance
Transitioning from an experimental sandbox to a validated environment requires a formal governance structure. Organizations must stop treating advanced tools as simple IT upgrades and start treating them as highly regulated assets. True governance rests on five core pillars.

Pillar 1: Use Case Intake and Risk Classification
You should not give every department open access to activate advanced tools whenever they want. A mature company implements a formal intake process. Before a single line of code is written or a vendor software is purchased, the business must capture the exact purpose, ownership, and expected benefit of the tool.
The tool must be classified by risk once captured. A helpful framework divides applications into three buckets:
High Risk: Systems that support clinical decision making, patient safety, quality control inspections, or deviation management. These require absolute validation rigor, design controls, and intense testing.
Medium Risk: Tools used for operational forecasting, supply chain streamlining, or trend analysis. These require clear procedural controls and standard validation.
Low Risk: Systems used for simple productivity, basic grammar corrections, or internal meeting scheduling. These require basic security reviews but minimal validation.
By tying your compliance controls directly to the risk level, you avoid over-documenting low-risk tools while assuring high-risk applications are bulletproof.
Pillar 2: Data Controls and ALCOA+ Principles
Every piece of information used by an advanced system must comply with strict data-integrity guidelines. This means all data must be attributable, legible, contemporaneous, original, and accurate. It must also be complete, consistent, enduring, and available.
Purpose-built solutions enforce these principles by creating strong data boundaries. They restrict the software so it can only access approved, verified source systems. They block the tool from pulling random information from the public internet. Furthermore, the system must keep an immutable audit trail. Every single prompt, every generated response, and every user validation must be permanently stamped with a time, date, and user identity.
Pillar 3: Mandatory Human Review
No advanced system should operate completely on autopilot when product quality or human lives are on the line. Governance frameworks ought to mandate a qualified human reviewer to check the work.
The software acts as an assistant, not the final judge. If the tool drafts a response to a quality deviation, a trained quality professional must review the source data, verify the accuracy of the draft, and officially sign off on the record. The system must store this human verification as part of the permanent compliance history.
Pillar 4: Continuous Performance Monitoring
Because advanced software can shift over time, you need a preemptive strategy to catch errors before they reach an auditor. This involves formulating clear metrics for model exactness, sensitivity, and fault rates.
Organizations must run regular challenge tests. These tests feed the system known data sets to verify that it still produces the expected results. If the performance drops below some threshold, it must trigger an automatic alert. The tool is then taken offline or restricted until a change control process evaluates the issue and revalidates the configuration.
Pillar 5: Thorough Vendor Qualification
Most companies do not build advanced language models from scratch. They partner with IT providers or integrate specialized software into their operations. However, regulators hold you responsible for your vendors' compliance.
You must thoroughly audit your technology partners. You need to inspect their security measures, bias detection protocols, and change control processes. If a vendor pushes an unannounced software update that alters how the model reasons, your validated status could vanish instantly. You must use vendors that offer complete honesty and give you control over when updates are applied.
Applying Computer Software Assurance to Advanced Systems
The thought of validating a dynamic, learning model can terrify traditional quality assurance teams. If you try to apply old, paperwork-heavy computer systems validation methods to advanced software, you will quickly find yourself buried in endless documentation. A typical project could take eight months to complete, destroying your competitive advantage.
Fortunately, the regulatory domain has evolved. The finalized computer software assurance guidance provides a modern framework that aligns perfectly with advanced technology.
Computer software assurance flips the script on validation. Instead of spending eighty percent of your time writing exhaustive test scripts and twenty percent on critical thinking, this system tells you to spend most of your time on risk analysis and critical thinking. It allows teams to focus their testing energy on the specific functions that directly impact product quality and patient safety.
When you apply this approach to advanced technology, validation becomes manageable. Instead of testing every likely response the tool could ever generate, you focus on the workflow's configuration. You test the boundaries, the data connectors, the human review steps, and the failure modes.
Organizations that utilize this risk-based framework see massive improvements. Validation timelines can drop from several months to just a matter of weeks. This allows life sciences companies to deploy powerful, automated solutions quickly without sacrificing a single shred of regulatory compliance.
How Purpose Built Compliance Platforms Solve the Problem
Living in the gap between a pilot and a validated system is dangerous and expensive. It wastes time, frustrates engineers, and leaves your business exposed to severe regulatory penalties. The solution is to step away from generic tools and adopt systems built from the ground up for regulated environments.
This is where specialized platforms make a massive difference. For instance, companies planning to manage their complex operations turn to dedicated provider ecosystems like PSC Software. Instead of trying to force a consumer application to comply with strict global laws, organizations leverage platforms designed with compliance as a core feature.
When you look at the product offerings within the life sciences ecosystem, you can see how purpose-built tools bridge the gap. For example, managing the intense training demands of a regulated workforce requires absolute precision. Neither a manual spreadsheet nor a standard corporate training tool can withstand the pressure of an audit. Using an automated option like the ACE LMS software solution ensures that every training event, standard operating procedure update, and employee qualification is tracked inside an unalterable audit trail. This level of control perfectly aligns with the information-consistency standards required for advanced automation.
Traditional validation paperwork can slow an organization to a crawl. Converting to a digital, paperless environment using tools like ACE Validation's paperless GxP software allows teams to unify their compliance activities. With document control, corrective actions, and validation records live in a single, connected digital ecosystem, implementing and monitoring advanced technology becomes simple, enabling effortless tracking of data lineage, transparent management of system changes, and a clear, organized history for any inspector who walks through your door.
A Practical Roadmap to Inspection Readiness
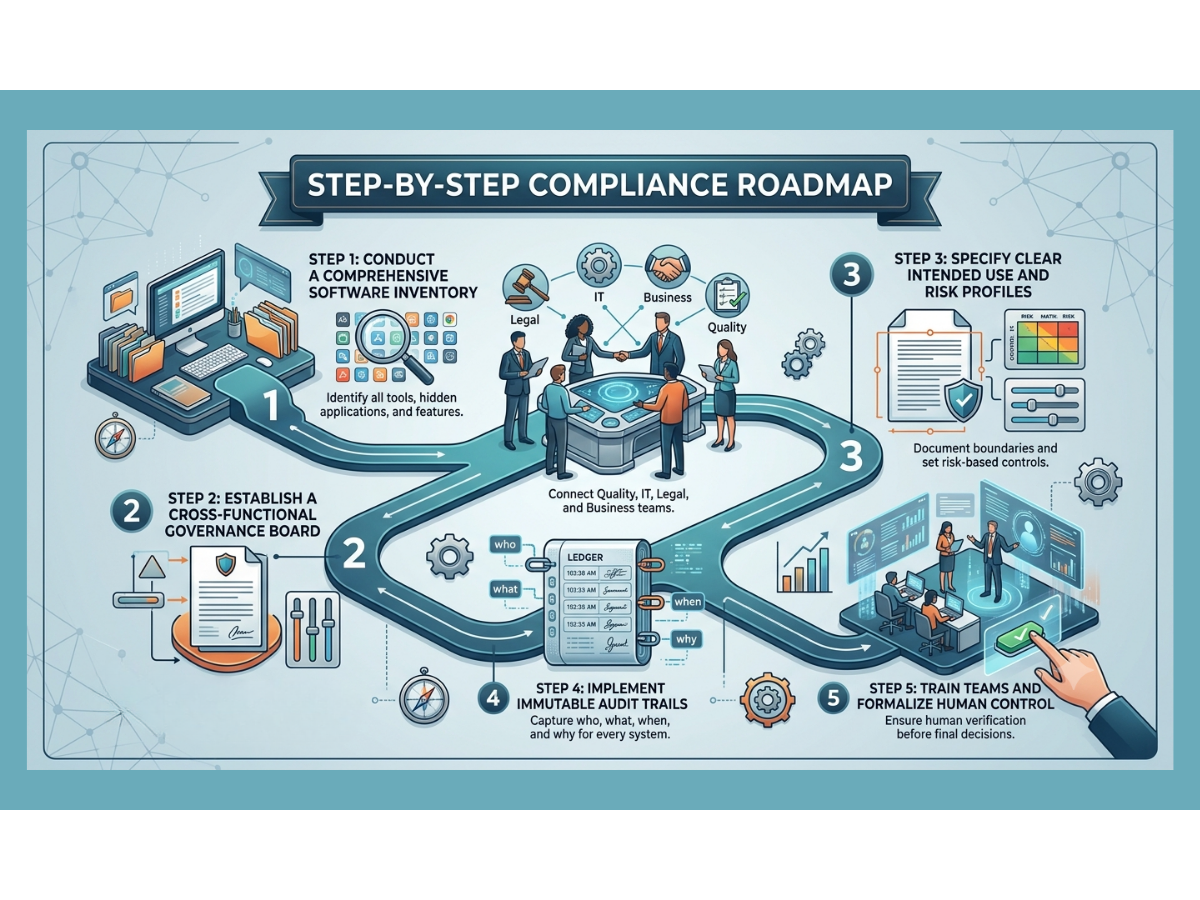
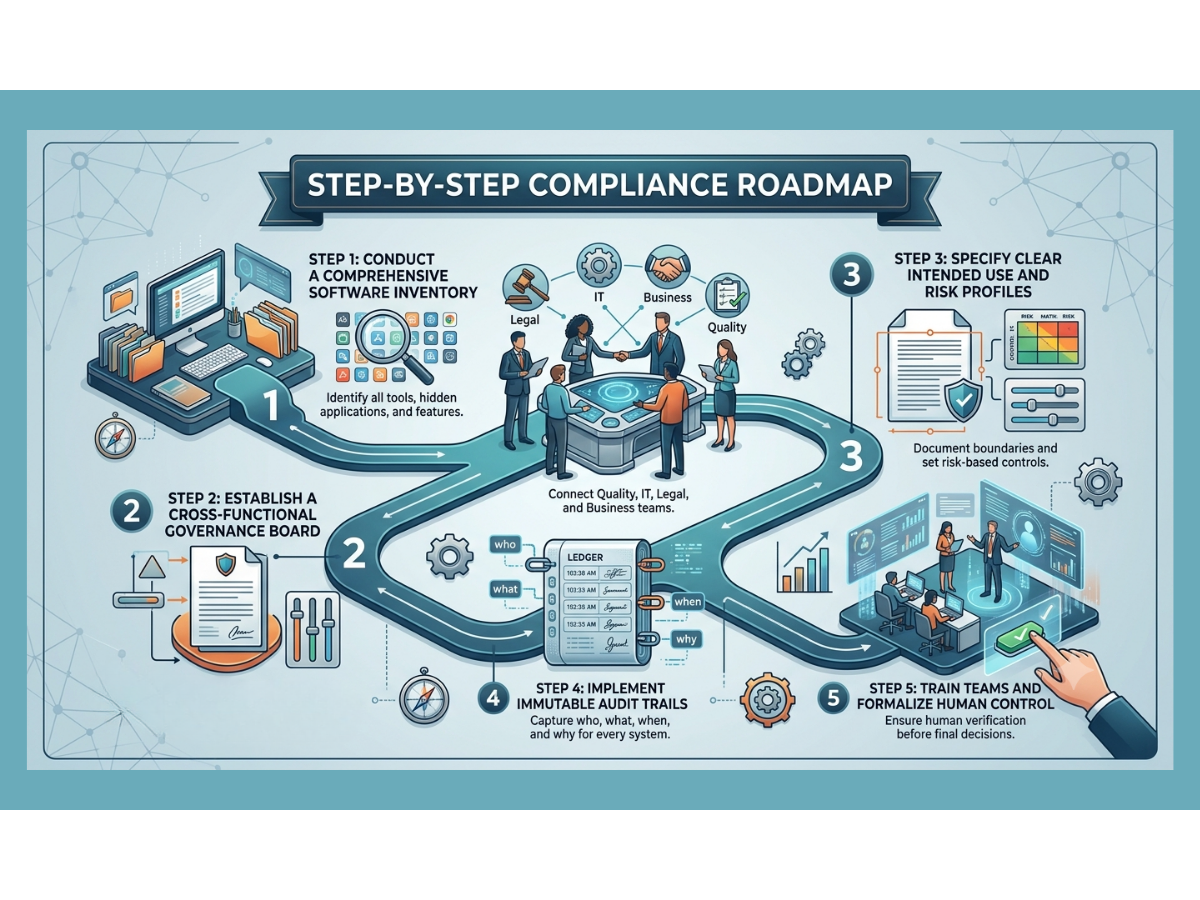
If your organization wants to close the gap and build artificial intelligence systems that are truly inspection-ready, you must follow a clear, step-by-step roadmap.

Step 1: Inventory All Advanced Tools
You cannot govern what you do not know exists. Conduct a thorough audit across your business to discover every tool currently in use. Look for hidden applications where employees might be pasting company data into public websites. Document every vendor-supplied feature that claims to use smart automation.
Step 2: Create an Internal Governance Board
Bring together leaders from quality assurance, information technology, legal, and operational business units. This group will serve as the gatekeepers for all automation projects. They will review new use cases, assign risk classifications, and confirm that no project moves forward without a clear validation plan.
Step 3: Draft Clear Intended Use Statements
For every approved tool, write a detailed statement explaining exactly what the software is allowed to do and what it is strictly prohibited from doing. Document the data sources, the human review workflows, and the exact records the system will generate.
Step 4: Enforce Technical Data Controls
Work with your IT team or software vendors to confirm that every system has strong access controls, data encryption, and unalterable audit trails. Verify that the system layout prevents automatic model updates without requiring formal change control.
Step 5: Establish Continuous Monitoring Standards
Create a schedule for regular performance reviews. Define your drift thresholds and write clear standard operating procedures for what the team must do if the software shows signs of declining accuracy.
Final Thoughts
Artificial intelligence offers incredible potential for the pharmaceutical industry and can help us analyze massive data sets, spot manufacturing deviations early, and streamline heavy documentation workloads. But these benefits mean absolutely nothing if the technology cannot survive a regulatory inspection.
The era of playing around with casual pilots is over. Regulatory bodies are stepping up enforcement, and the companies that succeed will be those that treat automation with the discipline it deserves. By shifting away from generic tools and deploying purpose-built software, sound risk frameworks, and complete data lineage, you can confidently move your technology out of the sandbox and into a fully validated, inspection-ready reality.
Bridge that gap between AI innovation and regulatory reality. Contact Metis Consulting Services today. We are experts who can streamline your computer software assurance, fortify your governance structure, and ensure your technology is fully validated and completely inspection-ready.

The Rise of Patient-Centric Packaging in Pharmaceuticals
In recent years, the pharmaceutical industry has begun to rethink its approach to drug packaging. No longer is packaging only a protective shell for medicines. Today, it is evolving into something much more meaningful: a bridge between drug makers and patients. This shift is called patient-centric packaging. In this post, we will explore what patient-centric packaging is, why it matters, and how it is transforming the way medicines are delivered and used.

This week in the Guardrail... we examine the fundamental shift occurring in pharmaceutical outsourcing as companies recognize that packaging is no longer just a container but a critical tool for improving patient adherence and safety.
By Michael Brofman, for Metis Consulting Services
Monday November 24, 2025
In recent years, the pharmaceutical industry has begun to rethink its approach to drug packaging. No longer is packaging only a protective shell for medicines. Today, it is evolving into something much more meaningful: a bridge between drug makers and patients. This shift is called patient-centric packaging. In this post, we will explore what patient-centric packaging is, why it matters, and how it is transforming the way medicines are delivered and used.
What Is Patient-Centric Packaging?
Patient-centric packaging means designing medicine packaging around the needs, abilities, and experiences of the people who will use it. According to Esko, a leader in packaging design, this kind of packaging considers three key elements: patient adherence, patient outcomes, and patient experience.
Traditionally, pharmaceutical packaging focused on safety, regulatory compliance, and product protection. But patient-centric design adds a new layer. It makes packaging more accessible, more intuitive, and more supportive of patients as they take their treatments.
Why Is This Shift Happening Now?
There are several reasons why pharmaceutical companies are embracing patient-centric packaging. Here are the main drivers:
Medication Adherence Problems
Many patients do not take their drugs exactly as prescribed. Poor adherence can lead to worse health outcomes and higher costs for the healthcare system.Aging Population
As more people grow older, there is a bigger need for packaging that is easy to open, read, and use. Many older patients have physical challenges, such as arthritis or reduced vision.Rise of Home Therapies
Treatments that were once administered in hospitals are now used at home, which includes biologics and injectables. For patients to self-manage safely, the packaging must help guide them.Trust and Safety Concerns
Patients need to know their medicines are authentic, safe, and appropriately stored. Innovative packaging helps build this trust.Digital Innovation
Technologies such as QR codes, NFC chips, RFID tags, and “smart labels” enable packaging to interact with the patient, provide information, and monitor use.Regulatory and Industry Pressure
Regulatory bodies and patient advocacy groups encourage more patient involvement in drug design, including packaging.Contract Packaging Growth
Pharmaceutical companies are outsourcing more packaging to experts who focus on patient-centric design.
What Does Patient-Centric Packaging Look Like?
Patient-centric packaging can take many forms. Here are some standard design features:
Blister Packs with Calendars
These are packs arranged by day and time so patients can clearly see when to take their medicine. For example, a 3 × 7 blister layout displays a three-week course in a single view.Multi-Compartment Containers / Pill Boxes
These let patients sort their medicine by dose. A meta-analysis shows that using these types of packaging improves adherence.Braille or Large-Print Labels
Some packaging provides accessibility for those with low vision or other challenges.Ergonomic Closures
Packaging that is easy to open, even for people with limited hand strength, is growing in demand.Smart Packaging
Innovative Packaging includes connected features: QR codes or NFC can link patients to digital leaflets, video instructions, or reminders.Serialization & Anti-Counterfeiting
Packaging can include RFID tags, tamper-evident seals, and other security features to assure patients that their drug is genuine.Sensor-Enabled Packaging
For temperature-sensitive medicines (such as biologics), packaging can include sensors that monitor storage conditions.
Real-World Examples
One well-known example of patient-centric packaging is ClearRx, a redesign of the standard medicine bottle created by designer Deborah Adler. The ClearRx bottle stands on its cap so the label folds over the top, which makes the drug name easy to see. The label also uses a large font, and there is a place for a color ring so different people in a household can tell their medicines apart.
Big pharmaceutical companies are also doing more. In several studies, companies have reported that patient-centered packaging makes medicine use more intuitive and self-explanatory.
Benefits of Patient-Centric Packaging
Why does patient-centric packaging matter? Here are some significant advantages:
Improves Adherence
By helping patients remember when and how to take their medication, patient-centric packaging supports better adherence.Reduces Errors
Clear instructions, intuitive layouts, and better labeling reduce the risk of misuse.Builds Trust
Innovative packaging features help patients verify authenticity and track storage conditions, building confidence in their treatment.Supports Accessibility
Packaging designs that consider older adults or people with disabilities make medications more straightforward to use.Enables Better Communication
Digital packaging can connect patients directly to educational content, helplines, or telehealth services.Helps Sustainability
More innovative packaging can reduce waste and support environmental goals, especially as the industry moves toward more sustainable materials.Regulatory Alignment
Innovative packaging helps companies meet regulatory requirements for serialization, traceability, and other requirements.
Challenges and Risks
While patient-centric packaging offers many benefits, it also comes with real challenges:
Cost
Designing and manufacturing new packaging solutions costs more than simply using traditional containers.Regulatory Burden
Changes to packaging must comply with strict regulations. Any redesign may require new approvals.Technology Adoption
Not all patients will use or trust digital features like QR codes or smart sensors. Some may lack smartphones or digital literacy.Supply Chain Complexity
Connected packaging and smart labels may require new logistics, serialization, and supply chain management.Privacy and Data Security
If packaging tracks use or transmits data, companies must protect patient privacy and secure their systems.Sustainability Trade-offs
While some innovative packaging is eco-friendly, others may require more materials or electronic components, which create waste.
What Is the Industry Doing to Overcome These Challenges?
Pharma companies, packaging firms, and contract manufacturers are working on solutions:
Outsourcing to Packaging Experts
Many drug makers are hiring contract packaging organizations that specialize in patient-focused designs.Engaging Patients Early
In some projects, companies talk to patients during development to learn what works best for them.Using Human-Factor Engineering
Designers apply what is known as “human factors” to make packaging more straightforward to use (for example, easier caps, larger print).Implementing Smart Technologies
Packaging developers are embedding NFC chips, QR codes, sensors, and serialization to bring packaging into the digital age.Developing Digital Information Services
Instead of relying solely on paper leaflets, companies are offering electronic patient information leaflets (ePIL) that users can access via smartphones.Improving Multi-Compartment Packaging
By building better blister packs, MDDS (multi-dose dispensing systems), and pill boxes, pharma companies are making it easier for patients to manage their regimen.Balancing Innovation and Sustainability
Firms are exploring sustainable materials while still adding innovative features.
What Does the Future Hold?
Looking ahead, patient-centric packaging is likely to become even more common. According to recent market forecasts, connected and intelligent packaging will continue to grow as key areas of innovation.
We can expect to see:
More personalized packaging tailored to individual patients (for example, dose-specific packets for personalized medicine).
Smart sensors that monitor conditions like temperature and communicate with patient apps or providers.
Multimedia support (video, audio) built into packaging to help patients understand how to take their medicines safely.
Greater regulatory support for patient-centered designs, especially as patient engagement becomes a priority in health care.
Sustainability integration, where eco-friendly materials align with patient safety and usability.
Why This Matters for Patients and Pharma
For patients, the rise of patient-centric packaging means better experiences, fewer mistakes, and more substantial confidence in their treatment. It may help people take their medicine correctly, avoid serious health risks, and live with more independence.
For pharmaceutical companies, focusing on patient-centric design is not only the right thing to do, it also makes good business sense. Better adherence means more effective therapies. Innovative packaging can reduce recalls, improve brand trust, and even open new opportunities for patient engagement.
Final Thoughts
The rise of patient-centric packaging marks a fundamental shift in how the pharmaceutical industry sees its role. Packaging is no longer just a box or a bottle. It is a key part of the patient journey. By designing packaging that is thoughtful, accessible, and smart, companies are placing patients at the center of their innovation.
This change is more than a trend; it is a movement toward safer, more effective, and more human care. As technology advances and patient voices grow stronger, we can expect packaging to become even more deeply rooted in meeting real-world patient needs.
The era of patient-centric packaging is here, demanding innovation, regulatory compliance, and a revamped supply chain. Don't let these complex challenges become a risk; contact Metis Consulting Services today at Hello@Metisconsultingservices to guide your team’s strategy, aligning your packaging design with human-factor engineering and digital technologies to capture value and ensure patient trust.
References
Esko. “Patient-Centered Packaging – Changing the Pharma Focus.” Esko.
Pharma Manufacturing. “Building stronger patient trust through packaging design.”
MDPI. “Patient Centric Pharmaceutical Drug Product Design: The Impact on Medication Adherence.”
Carli Lorenzini G, Olsson A. Exploring How and Why to Develop Patient-Centered Packaging: A Multiple-Case Study with Pharmaceutical Companies. Ther Innov Regul Sci. 2022 Jan;56(1):117-129. doi: 10.1007/s43441-021-00338-0. Epub 2021 Sep 28. PMID: 34581997; PMCID: PMC8688390.Röchling Medical. “Patient-Centric Pharmaceutical Packaging Design.”
CPHI Online. “2025 Pharmaceutical Packaging Market Prospects.”PubMed. “Exploring How and Why to Develop Patient-Centered Packaging: A Multiple-Case Study with Pharmaceutical Companies.” Lund University Publications.
GreyB. “Pharma Packaging: Top Challenges and Solutions in 2025.”
Exploring how and why to develop patient-centered packaging: A multiple-case study with pharmaceutical companies | Lund University Publications. https://lup.lub.lu.se/search/publication/3f185851-48e5-4929-9886-2b7ae69671f5

The Advantages of Bringing Pharmaceutical CMOs Back to the United States
Bringing CMOs back to the United States

For Metis Consulting Services, Inc.
By Michael Bronfman
August 11, 2025
This week in the "Guard Rail," we at Metis are exploring "Reshoring" of CMOs. We can't afford to settle for anything less than a fortified, domestic, and regional pharmaceutical industry. For decades, the lure of international manufacturing offered a path of lower costs, but this road has proven to be full of potholes.
The Benefits of Bringing Pharmaceutical CMOs Back to the United States (Reshoring)
The pharmaceutical industry plays a central and critical role in public health. Every stage in the drug development and manufacturing process impacts the final quality and safety of medicines. Contract Manufacturing Organizations, known as CMOs, are third-party organizations that manufacture drugs for pharmaceutical firms. These organizations handle activities ranging from producing active pharmaceutical ingredients (API) to packaging and labeling.
Over the past several decades, a large number of pharmaceutical manufacturers have moved overseas. Let's talk about why this is happening: cost savings, reduced labor expenses, and relaxed regulatory environments often tempt companies to China and India.
There have been growing discussions lately about the benefits of bringing pharmaceutical CMOs back to the United States. The term for this movement is "reshoring." The trend to shift overseas has come with a set of challenges and risks that directly impact quality, safety, and national security. Although reshoring requires investment of all kinds, including time and workforce development, among others, it also brings a wide range of returns on those investments. These advantages include improved supply chain resilience, increased product quality, strengthened national security, job creation, and a reduction in reliance on foreign manufacturing. And isn't that what we all want?
Here, we will take a bigger look at how reshoring CMOs to the United States offers long-term benefits to both the pharmaceutical industry and the public.
Improved Supply Chain Reliability
Pharmaceutical manufacturing operates most effectively with a stable supply chain. Delays, shortages, and disruptions have serious consequences for patients' access to the drugs they need. The complexity of global supply chains is in itself a challenge that creates multiple points of vulnerability. Drugs may pass through several countries before reaching their final destination. Disruption along this path, at any point, can lead to delays or stockouts. The long, complex chain is vulnerable to myriad forms of delay, including political tensions, natural disasters, or transportation failures.
By relocating CMOs to the United States, pharmaceutical companies can reduce the number of steps involved in the supply chains. As a result, we would expect faster delivery of finished products and improved response times during public health emergencies. A domestic manufacturing base allows for greater control over production scheduling and inventory management.
During the COVID-19 pandemic, global supply chain disruptions exposed the risks of overdependence on foreign manufacturing. Not just for us here in the US, but globally. Shortages of essential medications and active pharmaceutical ingredients were rampant. A more localized supply chain could help prevent similar problems in the future.
Enhanced Quality Control and Regulatory Oversight
The United States Food and Drug Administration enforces strict regulatory standards. The manufacturers must follow detailed guidelines to ensure safety, consistency, and efficacy. When pharmaceutical companies outsource production to overseas CMOs, consistent quality and quality oversight are more challenging. Regulatory agencies often lack the same reach and oversight capabilities in other countries.
If their CMOs are located back here in the United States, companies gain better access to real-time oversight, audits, inspections, and monitoring. Regulatory compliance is easier to enforce, and deviations from quality standards can be addressed more quickly. This results in fewer product recalls, improved batch consistency, and greater confidence in the quality of the medication supply.
Patients should always be the guiding light in pharmaceutical manufacturing. They deserve safe and effective treatments. A return to domestic production would enhance quality assurance. Improving it every step of the way, from raw material sourcing to final packaging.
Stronger National Security
Pharmaceutical products are a cornerstone of national health and security. When production is concentrated overseas, vulnerabilities become more apparent. Whether it is interruptions to supply or trade restrictions, or foreign political instability, we have more challenges to the health and security. In times of crisis, foreign governments may prioritize domestic needs and restrict exports of critical medications.
Now let's look at that risk when considering essential medications such as antibiotics, vaccines, and insulin. The lack of domestic manufacturing capacity limits the nation's ability to respond to emergencies. If there is another pandemic, or there are bioterrorism threats, or a natural disaster, reshoring pharmaceutical CMOs will strengthen national security by reducing dependence on international suppliers. This will allow for faster production of essential drugs in response to urgent needs. We need to mitigate the vulnerability before any of these disasters strike. With a domestic manufacturing infrastructure in place, as a result, the United States, or even the Americas, will be able to better protect its citizens during emergencies and avoid the harmful effects of drug shortages.
Economic Growth and Job Creation
Potential for economic development is another major advantage of bringing CMOs back to the United States. The pharmaceutical industry is a sector that is growing and expanding. This vital industry can provide high-paying jobs in science, engineering, quality control, and logistics.
Local communities are economically stimulated in related industries, including transportation, utilities, and construction. Building new manufacturing facilities or expanding existing ones could create employment opportunities for both skilled and entry-level workers. As more companies invest in domestic production, entire ecosystems develop around pharmaceutical hubs. These ecosystems create long-term economic benefits that go beyond the companies themselves.
In regions facing economic decline, pharmaceutical manufacturing plants have the potential to provide a much-needed economic boost. The jobs that are created tend to have better wages and benefits than many other industries, contributing to a higher standard of living. This, in turn, creates community stability.
Increased Transparency and Accountability
Patients, providers, and regulators must know where medications are produced and under what conditions. Transparency is essential. When production is moved overseas, transparency often decreases.
Domestic manufacturing encourages greater openness. Regulatory agencies have greater ease of access to inspect facilities and review records. Companies can communicate more clearly with the public about sourcing, safety, and compliance. This builds trust between the pharmaceutical industry and the patients it serves.
Consumers are showing interest in where their medications are made. Just as people care about the origin of their food, many want to know whether their medicines are produced safely and ethically. Reshoring supports this desire for greater accountability and corporate responsibility.
Technological Advancements and Innovation
When pharmaceutical manufacturing is brought back to the United States, there is a greater opportunity for innovation. Continuous manufacturing, advanced automation, and improved quality control systems are all more likely with a chain of domestic facilities. They are more likely to adopt cutting-edge technologies. These technologies increase efficiency, reduce costs over time, and enhance product consistency.
In contrast, many overseas facilities are slower to modernize due to limited capital investment or regulatory restrictions. Reshoring CMOs allows American firms to lead in pharmaceutical technology and manufacturing science.
Collaboration is strengthened: manufacturers, research institutions, and universities work together more naturally. The exchange of knowledge and technology accelerates innovation and shortens the time needed to bring new treatments to market.
Resilience in Times of Crisis
We have seen how vulnerable the global pharmaceutical supply chain can be. Recent events have led to delays, shortages, and rising prices. When companies rely too heavily on foreign suppliers, they lose the ability to adapt quickly to changing circumstances.
Creating a network of CMOs domestically increases resilience. Manufacturers will be able to launch emergency initiatives in a timely manner. They can adjust production levels or shift resources without waiting for overseas partners. This flexibility is essential during times of national crisis.
By investing in domestic capacity now, pharmaceutical companies can ensure they are prepared for the challenges of tomorrow. Reshoring is a long-term strategy that increases preparedness and stability.
Ethical and Environmental Considerations
Ethical labor practices and environmental standards can vary widely across different countries. CMOs may or may not operate under conditions that do not align with US values. There might be extremely low wages, unsafe working conditions, or limited environmental protections.
Bringing pharmaceutical manufacturing back to the US ensures compliance with fair labor laws and environmental regulations. Companies are required to provide safer working conditions and reduce their environmental impact. And consumers, in this case, patients, are increasingly interested in how products are made. These efforts support sustainability goals and improve corporate reputation.
Ethical sourcing and responsible production practices are no longer optional. Reshoring aligns with public expectations and supports the broader goal of corporate social responsibility.
I hope that after reading this, we all can agree that the decision to bring pharmaceutical CMOs back to the United States is both strategic and responsible. Offshore manufacturing has seemed to offer short-term cost savings. At the same time, it has created long-term risks related to quality, supply chain stability, and national security.
By investing in domestic production, the pharmaceutical industry can strengthen its foundation. Advantages include more reliable supply chains, enhanced quality control, stronger national security, economic growth, technological leadership, and ethical transparency.
Reshoring is certainly not without its challenges; it requires capital investment, workforce development, and regulatory planning. The long-term benefits do outweigh the initial costs. We can deliver safer, more reliable treatments to the people who need them most by producing more of our medications closer to home. And our industry will do all of that with a smaller footprint.
As the pharmaceutical industry faces growing complexity and rising public expectations, reshoring CMOs is a powerful step toward a more secure, transparent, and innovative future. The time has come to rebuild U.S. pharmaceutical manufacturing, for both economic reasons and the health and well-being of the nation.
If you are in a position to contract your organization's CMO and would like to discuss how to reshore manufacturing, please contact us at
hello@metisconsultingservices.com
Or for more information, see our website at:
https://www.metisconsultingservices.com/
Or better yet, schedule an appointment:
https://calendly.com/sbradley-metisconsultingservices

Navigating the Current Regulatory Climate
Here are some ways Metis Consulting Services could help your organization meet the challenges highlighted in a survey * of over 200 regulatory, safety, and quality directors from small to medium-sized biotechs in the EU (Ireland) and North America (U.S.)
Written by Michael Bronfman

Need help in the current Regulatory Climate? A consultant can help you get to Compliance and Beyond
To address the challenges faced by biotech companies as they enter new markets, a good consultant can leverage its expertise in navigating complex regulatory environments, ensuring compliance, and optimizing quality systems. Here are some ways Metis Consulting Services could help your organization meet the challenges highlighted in a survey * of over 200 regulatory, safety, and quality directors from small to medium-sized biotechs in the EU (Ireland) and North America (U.S.) :
1. Navigating Complex Quality Requirements
Metis Consulting can assist biotech companies in setting up and optimizing quality management systems (QMS) to meet the specific regulatory and licensing requirements of new markets. By providing expert guidance on local regulatory frameworks, Metis can help clients avoid common pitfalls related to misunderstanding market-specific requirements.
2. Pharmacovigilance Expertise
The survey highlighted pharmacovigilance as a critical challenge. Metis could offer tailored pharmacovigilance services to help biotech companies integrate and scale their pharmacovigilance activities early in the development process. This would include creating robust systems for adverse event reporting, risk management, and regulatory submissions to ensure compliance in various markets.
3. Streamlining Regulatory Approval Processes
With regulatory approval times being a significant challenge, Metis can help companies navigate regulatory submission and approval processes more efficiently. This can include regulatory strategy development, gap analysis, preparation of submissions, and liaison with regulatory bodies to expedite approvals and reduce delays.
4. Cost Management and Market Strategy
To mitigate the higher-than-expected costs and avoid market entry withdrawal, Metis could assist in conducting cost-benefit analyses for entering new markets. This would involve identifying the most cost-effective strategies for market entry, regulatory compliance, and operational scaling in challenging markets like China, Brazil, and others.
5. Early Integration of Key Planning Functions
The survey pointed out the need for early integration of pharmacovigilance, regulatory, and quality planning. Metis could offer integrated consulting services that help biotech companies align their regulatory, quality, and safety strategies from the outset. This proactive approach can help reduce the likelihood of unforeseen roadblocks and inefficiencies as companies scale and enter new markets.
6. Adjusting Strategies for the U.K. and EU Markets Post-Brexit
With heightened focus on the U.K. due to Brexit and the evolving regulatory landscape, Metis Consulting can offer specialized services to help biotech companies navigate post-Brexit regulatory challenges in the U.K. and the EU, ensuring that they remain compliant while maintaining smooth operations across both markets.
7. Success Redefined: Compliance and Investor Relations
As compliance and investor payback become more critical success metrics, Metis Consulting can help biotech companies develop robust compliance strategies that align with the increasingly stringent requirements in the U.S. and other markets. Additionally, Metis could assist companies in crafting strategic plans to demonstrate strong compliance performance, which would be crucial for maintaining investor confidence.
8. Strategic Market Entry and Representation
For companies looking to expand into high-priority markets such as the U.S., U.K., Canada, Brazil, and the Middle East, Metis can provide strategic advice on market entry, representation models (direct vs. partnerships), and regulatory considerations specific to those regions. This could include helping biotech companies decide whether to pursue direct representation or rely on local partnerships, depending on the market's dynamics.
9. Tailored Regional Guidance
Metis can also offer tailored insights into specific regions that are becoming increasingly important for biotech market access. For example, the firm could assist in navigating regulatory complexities in Brazil, which has become a key biotech market in Latin America, or help companies looking to expand into the Middle East, where the biotech sector is growing rapidly.
By offering these comprehensive consulting services, Metis Consulting can position itself as a trusted partner that helps biotech companies overcome regulatory, safety, and quality challenges while successfully entering and expanding in new markets."
* https://www.linkedin.com/pulse/top-pharma-biotech-news-1-january-edition-symmetrictraining-ndtgf/