
Quality Management System Regulation

By Michael Bronfman
June 15, 2026
Imagine a large medical factory building complicated machinery like heart pacemakers or robotic surgical arms. For close to thirty years, if that company wanted to sell its products inside the United States, they had to follow a specific book of government safety rules. If they also wanted to sell those exact same medical tools in Europe, Canada, or Japan, they had to open an entirely separate set of books to satisfy international rules. Engineers and quality inspectors spent thousands of hours filling out two different piles of paperwork for the exact same medical device.
This double system caused massive confusion and slowed down the arrival of life-saving technology to hospitals. This waste of energy finally came to an end on February 2, 2026. On that historic day, the United States Food and Drug Administration officially changed the law. They threw away their old factory rulebook and adopted the primary international standard used by the rest of the civilized world.
This massive shift is called the Quality Management System Regulation. It completely overwrites the older rules that medical engineers had memorized for decades. By taking the global gold standard for medical manufacturing and making it the official law of the land, the United States government has transformed how medical tools, biotech gear, and drug delivery systems are designed and built.
What is the New Rule for Device Makers
To truly understand this event, you have to look at the specific codes that govern medical manufacturing. For generations, the old rules lived inside a document known as 21 CFR Part 820. This was the traditional American Quality System Regulation. While it kept patients safe, it used unique language and isolated requirements that did not match the rest of the planet.
The new law unifies these separate worlds. The government accomplished this through a legal process called incorporation by reference. Instead of writing hundreds of pages of brand new American text, the Food and Drug Administration simply stated that the official global standard is now the foundational law of the United States.
The global standard they adopted is named ISO 13485. This is an international agreement created by manufacturing experts from dozens of countries. The specific details of this global transition can be explored directly on the Official FDA Quality Management System Regulation Page, which hosts the formal announcements and legal text. By linking American law directly to this global framework, a company can now design a single quality system that satisfies regulators in Washington, London, Tokyo, and Paris all at the exact same time.
The Death of the Old System and Birth of the New
This policy change is not a minor update or a cosmetic face lift. It is a complete structural teardown of the old regulatory architecture. The government has withdrawn the vast majority of the old text that factories used to build their inspection programs.
[Old System: 21 CFR Part 820] ──> Gaps Closed ──> [New System: QMSR] ▲ │ [Global Standard: ISO 13485]
This rewrite introduces major operational changes:
The Old Title is Gone: The phrase Quality System Regulation has been replaced by Quality Management System Regulation, signaling a shift toward total management responsibility.
The Inspection Method has Shifted: For decades, government inspectors used a tool called the Quality System Inspection Technique to grade factories. That manual has been retired completely.
New Audit Rules Apply: Inspectors now utilize an updated compliance program labeled 7382.850, which matches international factory audit methods.
No More Golden Exceptions: Under the older rules, companies could keep their internal management review notes and supplier audits private from everyday investigators. Under the new law, that shield has been dropped, and regulators have full access to those high level self evaluations.
How This Shifts the Focus to Holistic Risk Management
The most vital conceptual change in this new era is how companies must handle risk. In the old American system, risk analysis was treated like a single step in the blueprint phase. Engineers would brainstorm what might go wrong with a device before they built it, write a safety report, and then check that box off their list.
The international model changes that completely. It requires a holistic risk management approach. Under this framework, safety analysis is not a single event; it is a continuous loop that must influence every single corner of the factory throughout the entire life cycle of the product.
[Supplier Evaluation] ──> [Manufacturing Process] ──> [Customer Complaints] │ │ │ ▼ ▼ ▼ [Continuous Risk Review] ──> [Design Updates] ──> [Continuous Risk Review]
This means managers must look at risks when they choose a battery supplier, when they train a new assembly line worker, and when they review customer phone calls. If a machine detects a small flaw in a part on the factory floor, management cannot just replace the part. They must mathematically calculate if that small flaw could ripple outward and affect patient health months down the road. Every decision made inside the company must be driven by data and focused on minimizing risk to the end user.
The Massive Impact on Combination Products
While this transition is a big deal for pure device companies like wheelchair or scalpel manufacturers, it is an even bigger deal for biotech firms that create combination products. A combination product is a medical asset that merges a drug, a device, or a biological product into one single package.
[Drug Component] (Liquid Asthma Medicine) + ──> [Combination Product] (Inhaler) [Device Component] (Plastic Spray Nozzle)
Think about a modern insulin pen, a pre filled medicine syringe, or a high tech asthma inhaler. These products are incredibly complex because they must follow both drug manufacturing laws and device manufacturing laws simultaneously. The rules for coordinating these dual systems live inside an official regulation called 21 CFR Part 4.
To prevent this transition from breaking the biotech industry, the government made careful conforming edits to these combination product laws. If a biotech company uses a device-centered quality system to build their inhaler, they must now prove they meet the new international standard while still satisfying specific drug testing laws, such as verifying the purity of the liquid medicine. To review how these intersecting guidelines function under the new law, developers can consult the FDA Quality Management System Regulation FAQ Document for direct clarity on how these systems overlap.
Navigating the Gaps Between International and Domestic Law
Even though the United States has adopted the international standard, there are still a few unique American legal requirements that the global rulebook does not cover. The global standard is designed for any country, but the United States has specific acts of Congress that cannot be overridden by an international committee.
To solve this problem, the government built a hybrid framework. The new law incorporates the global standard but inserts special clauses to close the remaining gaps. For instance, American laws regarding medical device tracking, unique device identification stickers, and formal reports of product recalls still remain fully active.
[The New Unified QMSR Framework] │ ┌──────────────────────────┴──────────────────────────┐ ▼ ▼[ISO 13485 Core Rules] [FDA Specific Additions]- Continuous Risk Management - Unique Device Tracking (UDI)- Global Vendor Audits - Formal Recall Reports- Executive Quality Oversight - Strict Patient Privacy Control
If an international clause ever conflicts with a specific text inside the Federal Food, Drug, and Cosmetic Act, the American law wins the argument. Manufacturers must be careful not to assume that a standard international certificate means they are completely safe from an American inspection. They must ensure their factory systems cover both the core international rules and the extra American additions.
Step-by-Step Implementation Plan for Manufacturers
For companies that have not yet fully transitioned their facilities to match this global overhaul, the path forward requires a methodical blueprint. Industry trade groups and government compliance officers suggest a four-step strategy to modernize quality systems without disrupting active assembly lines.
Conduct a Comprehensive Gap Analysis. Requires cross-department review. Compare every current manufacturing procedure against the specific clauses of the international standard to find blind spots, particularly around corporate management responsibility and vendor risk screening.
Rebuild Internal Quality System Architecture, updating core blueprints. Rewrite standard operating manuals to remove outdated terminology and integrate continuous risk assessment protocols into everyday purchasing, engineering, and shipping tasks.
Establish Expanded Supplier Monitoring Programs. Evaluate the outside supply chain. Create strict evaluation scorecards for third-party vendors, ensuring that the factory can audit component creators and trace the safety records of raw materials back to their source.
Execute Enterprise-Wide Compliance Training, Building a culture of safety, Train factory floor operators, line inspectors, and executive managers on the new standard, establishing a workplace culture where every employee actively looks for and reports hidden product risks.
The Broader Impact on Biotech Supply Chains
This regulatory shift ripples far beyond the walls of the primary medical device factory. It fundamentally alters the relationship between device makers and their outside component suppliers. Under the old system, if a company bought a plastic tube or a digital sensor from an outside vendor, they often just tested the part when it arrived at the warehouse door.
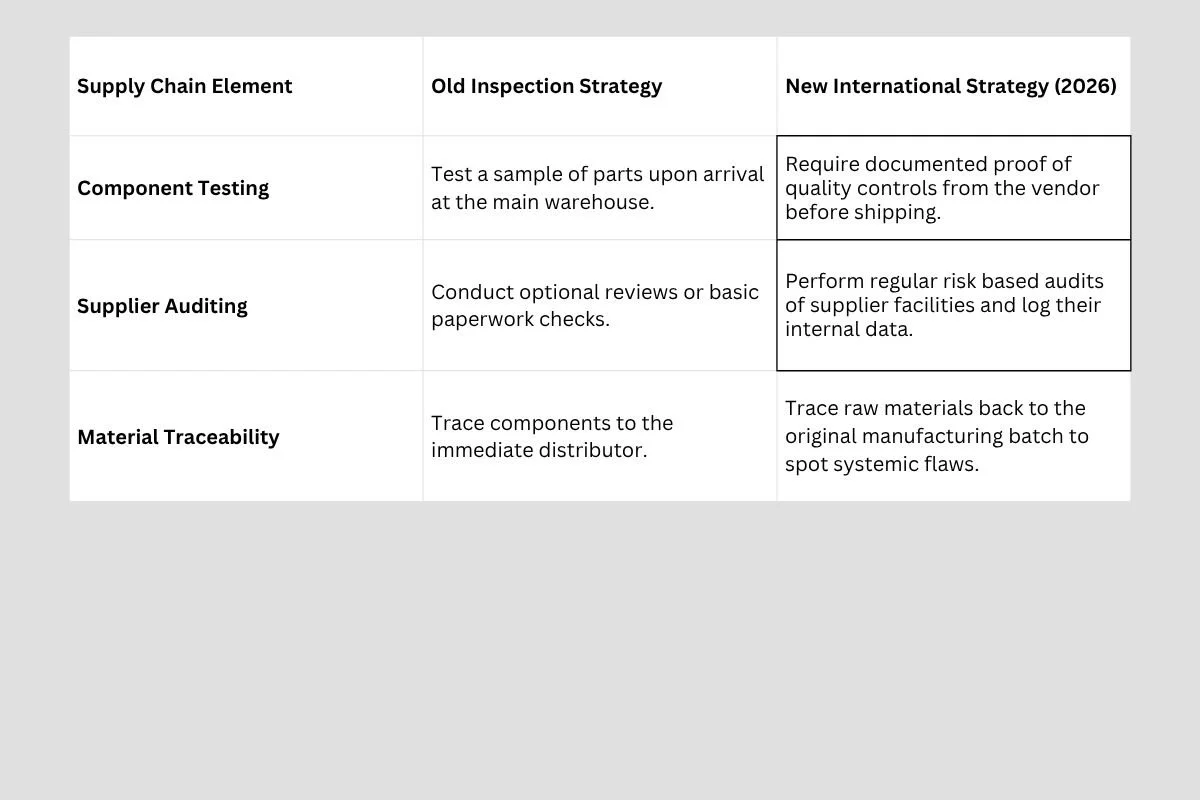
The new international focus requires extreme control over the entire supply chain. Device makers are now legally responsible for the quality systems of their suppliers. If a biotech firm buys an active electronic chip for a modern diagnostic machine, they must evaluate the chip maker's ability to maintain clean facilities and steady standards.
The new international focus requires extreme control over the entire supply chain. Device makers are now legally responsible for the quality systems of their suppliers. If a biotech firm buys an active electronic chip for a modern diagnostic machine, they must evaluate the chip maker's ability to maintain clean facilities and steady standards. This means component suppliers who want to work with major medical device firms must upgrade their own operations. Small machine shops and digital sensor makers must adopt rigorous tracking habits if they want to remain part of the global healthcare economy.
The Long Term Benefits for Patients and Industry
While this quality system overhaul demands significant upfront investment, training time, and software updates, the long-term benefits for global health are immense. By clearing away conflicting regulations, the industry can redirect massive amounts of capital from administrative paperwork directly into laboratory research and development.
For patients, this means that groundbreaking health tech developed anywhere on Earth can move through the regulatory evaluation process faster than ever before. A smart hospital monitor built in Germany can now be introduced to American clinics without months of administrative delays.
Furthermore, because the new system forces companies to look at risk continuously, the medical tools arriving at patient bedsides will be inherently safer and more resilient. The transition to this unified system marks a triumph for common-sense regulation. It proves that the global medical community can cooperate across borders to build a streamlined, safe, and innovative future for human care.
Ready to Navigate the New Era of Medical Device Compliance?
Streamline your global operations, and confidently accelerate your life-saving technologies to market.
Don't let compliance gaps slow down your innovation; contact Metis Consulting Services today