

Quality Management System Regulation

By Michael Bronfman
June 15, 2026
Imagine a large medical factory building complicated machinery like heart pacemakers or robotic surgical arms. For close to thirty years, if that company wanted to sell its products inside the United States, they had to follow a specific book of government safety rules. If they also wanted to sell those exact same medical tools in Europe, Canada, or Japan, they had to open an entirely separate set of books to satisfy international rules. Engineers and quality inspectors spent thousands of hours filling out two different piles of paperwork for the exact same medical device.
This double system caused massive confusion and slowed down the arrival of life-saving technology to hospitals. This waste of energy finally came to an end on February 2, 2026. On that historic day, the United States Food and Drug Administration officially changed the law. They threw away their old factory rulebook and adopted the primary international standard used by the rest of the civilized world.
This massive shift is called the Quality Management System Regulation. It completely overwrites the older rules that medical engineers had memorized for decades. By taking the global gold standard for medical manufacturing and making it the official law of the land, the United States government has transformed how medical tools, biotech gear, and drug delivery systems are designed and built.
What is the New Rule for Device Makers
To truly understand this event, you have to look at the specific codes that govern medical manufacturing. For generations, the old rules lived inside a document known as 21 CFR Part 820. This was the traditional American Quality System Regulation. While it kept patients safe, it used unique language and isolated requirements that did not match the rest of the planet.
The new law unifies these separate worlds. The government accomplished this through a legal process called incorporation by reference. Instead of writing hundreds of pages of brand new American text, the Food and Drug Administration simply stated that the official global standard is now the foundational law of the United States.
The global standard they adopted is named ISO 13485. This is an international agreement created by manufacturing experts from dozens of countries. The specific details of this global transition can be explored directly on the Official FDA Quality Management System Regulation Page, which hosts the formal announcements and legal text. By linking American law directly to this global framework, a company can now design a single quality system that satisfies regulators in Washington, London, Tokyo, and Paris all at the exact same time.
The Death of the Old System and Birth of the New
This policy change is not a minor update or a cosmetic face lift. It is a complete structural teardown of the old regulatory architecture. The government has withdrawn the vast majority of the old text that factories used to build their inspection programs.
[Old System: 21 CFR Part 820] ──> Gaps Closed ──> [New System: QMSR] ▲ │ [Global Standard: ISO 13485]
This rewrite introduces major operational changes:
The Old Title is Gone: The phrase Quality System Regulation has been replaced by Quality Management System Regulation, signaling a shift toward total management responsibility.
The Inspection Method has Shifted: For decades, government inspectors used a tool called the Quality System Inspection Technique to grade factories. That manual has been retired completely.
New Audit Rules Apply: Inspectors now utilize an updated compliance program labeled 7382.850, which matches international factory audit methods.
No More Golden Exceptions: Under the older rules, companies could keep their internal management review notes and supplier audits private from everyday investigators. Under the new law, that shield has been dropped, and regulators have full access to those high level self evaluations.
How This Shifts the Focus to Holistic Risk Management
The most vital conceptual change in this new era is how companies must handle risk. In the old American system, risk analysis was treated like a single step in the blueprint phase. Engineers would brainstorm what might go wrong with a device before they built it, write a safety report, and then check that box off their list.
The international model changes that completely. It requires a holistic risk management approach. Under this framework, safety analysis is not a single event; it is a continuous loop that must influence every single corner of the factory throughout the entire life cycle of the product.
[Supplier Evaluation] ──> [Manufacturing Process] ──> [Customer Complaints] │ │ │ ▼ ▼ ▼ [Continuous Risk Review] ──> [Design Updates] ──> [Continuous Risk Review]
This means managers must look at risks when they choose a battery supplier, when they train a new assembly line worker, and when they review customer phone calls. If a machine detects a small flaw in a part on the factory floor, management cannot just replace the part. They must mathematically calculate if that small flaw could ripple outward and affect patient health months down the road. Every decision made inside the company must be driven by data and focused on minimizing risk to the end user.
The Massive Impact on Combination Products
While this transition is a big deal for pure device companies like wheelchair or scalpel manufacturers, it is an even bigger deal for biotech firms that create combination products. A combination product is a medical asset that merges a drug, a device, or a biological product into one single package.
[Drug Component] (Liquid Asthma Medicine) + ──> [Combination Product] (Inhaler) [Device Component] (Plastic Spray Nozzle)
Think about a modern insulin pen, a pre filled medicine syringe, or a high tech asthma inhaler. These products are incredibly complex because they must follow both drug manufacturing laws and device manufacturing laws simultaneously. The rules for coordinating these dual systems live inside an official regulation called 21 CFR Part 4.
To prevent this transition from breaking the biotech industry, the government made careful conforming edits to these combination product laws. If a biotech company uses a device-centered quality system to build their inhaler, they must now prove they meet the new international standard while still satisfying specific drug testing laws, such as verifying the purity of the liquid medicine. To review how these intersecting guidelines function under the new law, developers can consult the FDA Quality Management System Regulation FAQ Document for direct clarity on how these systems overlap.
Navigating the Gaps Between International and Domestic Law
Even though the United States has adopted the international standard, there are still a few unique American legal requirements that the global rulebook does not cover. The global standard is designed for any country, but the United States has specific acts of Congress that cannot be overridden by an international committee.
To solve this problem, the government built a hybrid framework. The new law incorporates the global standard but inserts special clauses to close the remaining gaps. For instance, American laws regarding medical device tracking, unique device identification stickers, and formal reports of product recalls still remain fully active.
[The New Unified QMSR Framework] │ ┌──────────────────────────┴──────────────────────────┐ ▼ ▼[ISO 13485 Core Rules] [FDA Specific Additions]- Continuous Risk Management - Unique Device Tracking (UDI)- Global Vendor Audits - Formal Recall Reports- Executive Quality Oversight - Strict Patient Privacy Control
If an international clause ever conflicts with a specific text inside the Federal Food, Drug, and Cosmetic Act, the American law wins the argument. Manufacturers must be careful not to assume that a standard international certificate means they are completely safe from an American inspection. They must ensure their factory systems cover both the core international rules and the extra American additions.
Step-by-Step Implementation Plan for Manufacturers
For companies that have not yet fully transitioned their facilities to match this global overhaul, the path forward requires a methodical blueprint. Industry trade groups and government compliance officers suggest a four-step strategy to modernize quality systems without disrupting active assembly lines.
Conduct a Comprehensive Gap Analysis. Requires cross-department review. Compare every current manufacturing procedure against the specific clauses of the international standard to find blind spots, particularly around corporate management responsibility and vendor risk screening.
Rebuild Internal Quality System Architecture, updating core blueprints. Rewrite standard operating manuals to remove outdated terminology and integrate continuous risk assessment protocols into everyday purchasing, engineering, and shipping tasks.
Establish Expanded Supplier Monitoring Programs. Evaluate the outside supply chain. Create strict evaluation scorecards for third-party vendors, ensuring that the factory can audit component creators and trace the safety records of raw materials back to their source.
Execute Enterprise-Wide Compliance Training, Building a culture of safety, Train factory floor operators, line inspectors, and executive managers on the new standard, establishing a workplace culture where every employee actively looks for and reports hidden product risks.
The Broader Impact on Biotech Supply Chains
This regulatory shift ripples far beyond the walls of the primary medical device factory. It fundamentally alters the relationship between device makers and their outside component suppliers. Under the old system, if a company bought a plastic tube or a digital sensor from an outside vendor, they often just tested the part when it arrived at the warehouse door.
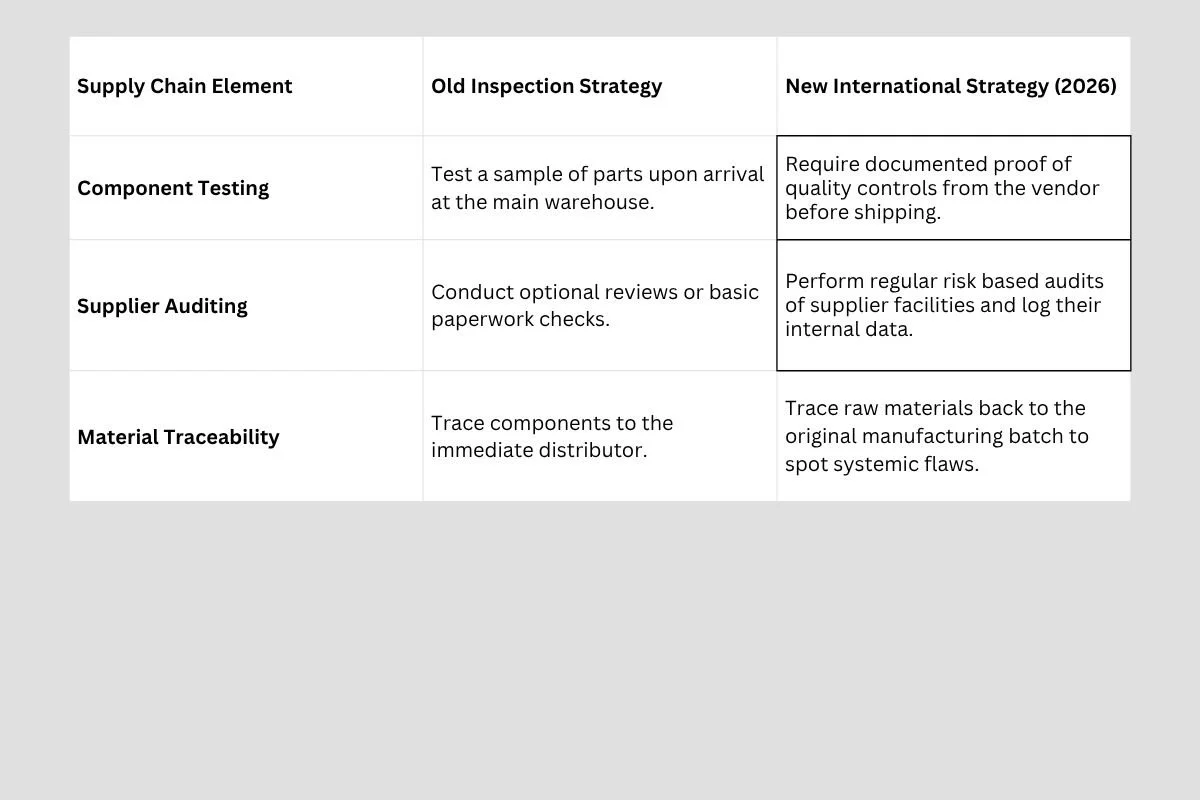
The new international focus requires extreme control over the entire supply chain. Device makers are now legally responsible for the quality systems of their suppliers. If a biotech firm buys an active electronic chip for a modern diagnostic machine, they must evaluate the chip maker's ability to maintain clean facilities and steady standards.
The new international focus requires extreme control over the entire supply chain. Device makers are now legally responsible for the quality systems of their suppliers. If a biotech firm buys an active electronic chip for a modern diagnostic machine, they must evaluate the chip maker's ability to maintain clean facilities and steady standards. This means component suppliers who want to work with major medical device firms must upgrade their own operations. Small machine shops and digital sensor makers must adopt rigorous tracking habits if they want to remain part of the global healthcare economy.
The Long Term Benefits for Patients and Industry
While this quality system overhaul demands significant upfront investment, training time, and software updates, the long-term benefits for global health are immense. By clearing away conflicting regulations, the industry can redirect massive amounts of capital from administrative paperwork directly into laboratory research and development.
For patients, this means that groundbreaking health tech developed anywhere on Earth can move through the regulatory evaluation process faster than ever before. A smart hospital monitor built in Germany can now be introduced to American clinics without months of administrative delays.
Furthermore, because the new system forces companies to look at risk continuously, the medical tools arriving at patient bedsides will be inherently safer and more resilient. The transition to this unified system marks a triumph for common-sense regulation. It proves that the global medical community can cooperate across borders to build a streamlined, safe, and innovative future for human care.
Ready to Navigate the New Era of Medical Device Compliance?
Streamline your global operations, and confidently accelerate your life-saving technologies to market.
Don't let compliance gaps slow down your innovation; contact Metis Consulting Services today

GLP and GCP: How the FDA and EPA Watch Over Science
Two of the most important sets of rules are called GLP and GCP. In 2026, the two main government agencies responsible for these rules, the Food and Drug Administration (FDA) and the Environmental Protection Agency (EPA), are working together more than ever before. Re: FDA & EPA Oversight.

This week in the Guardrail…
We explore how federal oversight is shifting standards in 2026, making rigorous data integrity the new baseline for every laboratory and clinic.
By Michael Bronfman
The world of making new medicines and chemicals is a very busy place. Every day, scientists are working in labs and clinics to find the next big cure or a safer way to clean our homes. Because these products can affect our health and the planet, the government has very strict rules to ensure everything is done correctly. Two of the most important sets of rules are called GLP and GCP. In 2026, the two main government agencies responsible for these rules, the Food and Drug Administration (FDA) and the Environmental Protection Agency (EPA), are working together more than ever before. This “shaking out” of the rules is changing how companies operate.
What Do These Letters Mean
To understand the future of science, you first have to know what these abbreviations stand for. They are like a specialized language for safety and honesty.
GLP stands for Good Laboratory Practice. These rules are for the early stages of research. This is the work that happens in a lab with test tubes, plants, or animals before a human ever touches the product. The Environmental Protection Agency uses these rules to ensure that a new pesticide or powerful cleaner will not harm the environment or the people using it.
GCP stands for Good Clinical Practice. These rules start when the research moves into a clinic and involves human volunteers. The Food and Drug Administration uses GCP to ensure that participants in medical studies are safe and that the results are truthful.
The FDA and the EPA Joining Forces
In the past, these two agencies mostly stayed in their own separate worlds. If a company were making a heart medicine, they would talk to the FDA. If they were making a new bug spray, they would talk to the EPA. But today, many new products fall into both categories. For example, a special soap that kills germs on your skin might be considered both a medicine and a chemical.
Because of this, the FDA and EPA are now sharing their notes. If an EPA inspector finds that a lab is messy or that the scientists are not recording their results correctly, they report it to the FDA. This means companies can no longer be “half good.” They have to follow the rules perfectly for both agencies. This coordination ensures that, no matter what kind of product is being made, the public is protected by the highest standards.Official Federal Register : How these agencies work together
Why Honesty Is the Only Policy
The main goal of both GLP and GCP is to ensure data integrity. Data is just a fancy word for the information and results gathered during an experiment. If a scientist says that a drug worked on ten people, the government wants to see the actual signatures and blood test results to prove it.
If a company is caught lying about its data, it can be fined millions of dollars. They might even be banned from ever making products again. This is why risk management is so important during the middle stages of a study. If a company finds a minor mistake in its lab records, it needs to fix it immediately. Waiting until later to “clean up” the paperwork is a huge risk that can lead to a total failure when the FDA or EPA comes to visit.
Protecting the People in the Studies
While GLP protects the science in the lab, GCP protects the people in the clinics. Every person who joins a clinical trial is a volunteer. They are doing something brave to help others. GCP rules ensure that these volunteers are treated with respect.
Under these rules, every volunteer must sign a form acknowledging the risks. This is called informed consent. The doctors must also closely monitor the volunteers for any side effects. If a patient experiences a headache or dizziness, it must be recorded in the official files. TheNational Institutes of Health provides extensive information on how these rules help keep people safe in medical research.
The Quality Control Revolution
In 2026, many companies are hiring specialized teams just to ensure quality. These teams are like the “referees” of science. They do not do the experiments themselves. Instead, they monitor the other scientists to ensure they follow all GLP and GCP rules.
They check that lab machines are properly calibrated. They check to ensure that every signature on a form is genuine and dated correctly. This might sound like a lot of extra work, but it saves the company from failing an inspection. When a company has a high “quality score,” the FDA and EPA can trust their results much more easily. Organizations like theSociety of Quality Assurance help train these specialized workers to stay up to date on the latest rules.
Using Technology to Stay Safe
Technology is making it easier for the FDA and EPA to do their jobs. In the old days, inspectors had to look through thousands of paper files. Now, most of the data is digital. This allows the government to look at the data in real time.
If a lab in California runs a test, an official in Washington, D.C., can see the results almost instantly. This helps catch mistakes before they become big problems. It also makes it harder for anyone to change their results later to make a drug look better than it really is. This transparency is a big part of why the “shake out” between the two agencies is happening so fast. Digital tools make it impossible to hide in the shadows.
The Global Impact of These Rules
Australia, Europe, and the United States all have their own versions of these rules. However, they are all starting to look very similar. This is good news for the public. It means that a drug tested in Australia can be sold in America as long as it follows the same high standards of GLP and GCP.
When countries agree on the rules, medicines can travel around the world much faster. This helps people in every country get the help they need without waiting years for additional testing. TheWorld Health Organization works to help all countries follow these same high standards for health and safety.
Education Is the Key
For these rules to work, every person in the pharmaceutical industry needs to be educated. It is not just the boss's job. Even the person cleaning the lab or the nurse at the clinic needs to understand why the rules matter.
Education helps people understand that following the rules is about more than just avoiding a fine. It is about making sure that the medicine your grandmother takes, or the soap you use on your children, is 100 percent safe. When everyone knows the “why” behind the rules, they are much more likely to follow them correctly every single day.
Risk Management and Quality Systems
Modern pharmaceutical companies use a Quality Management System (QMS) to track everything. A QMS is like a giant digital brain that stores all the company's rules and records. It helps with risk handling by flagging errors the moment they happen.
In 2026, risk management strategies are no longer just about fixing problems. They are about predicting them. By using clinical data management tools, a biotech firm can detect whether a machine is starting to wear out or whether a lab is experiencing too many small errors. This type of risk management planning helps prevent major disasters that lead to FDA warning letters.
The Role of REMS in Safety
Another way the FDA keeps people safe is through REMS, which stands for Risk Evaluation and Mitigation Strategies. These are extra safety programs for drugs that might be dangerous if not used exactly right. For example, some medicines require a patient to get a blood test every month. TheFDA REMS website tracks these programs to ensure drug companies are doing their part to manage risk.
Final Thoughts on the Future of Quality
The “shaking out” of rules between the FDA and the EPA is a positive step for everyone. It means that science is becoming more open and more honest. Companies are learning that they cannot take shortcuts during any stage of research.
By following the rules of GLP and GCP, we ensure that the future of medicine is bright. We can trust the products we buy because we know the government is watching and the scientists are being careful. Quality is not just a set of letters; it is a promise to the public that their safety is the most important thing of all. Don’t wait for a problem to appear before you start being careful. Start with quality on day one and the rest of the journey will be much smoother for everyone.
Frequently Asked Questions: GLP, GCP, and Regulatory Compliance
What is the main difference between GLP and GCP?The biggest difference is when they are used. GLP is for the preclinical stage, which is work done in a lab on animals or cells. GCP is for the clinical stage involving human volunteers.
Why does the EPA care about laboratory rules?The EPA ensures that chemicals like weed killers do not pollute our water. They use GLP to ensure companies are honest about chemical safety. You can find guidelines on theEPA Compliance Monitoring page.
Can a company fail a trial if they follow the science but miss the paperwork?Yes. In the eyes of the FDA, if a test was not documented correctly, it never happened. “Clean data” is just as important as the medicine itself.
What happens during an FDA or EPA inspection?Inspectors check original notebooks, computer logs, and even equipment logs. They want to see a clear trail from the study's start to the final report.
How does Risk Mitigation help with these rules?It means finding small mistakes before the government does. Fixing a mistake early in a study costs much less than fixing it later, when thousands of people are involved.
Where can I stay updated on these changing rules in 2026?The best place to watch for updates is theFDA Voice blog. This site tracks how the agencies are joining their rules for digital data. https://www.fda.gov/news-events/fda-newsroom/fda-voices.
The Pre-Inspection Compliance Checklist
When an inspector arrives, they look for a “culture of quality.” If you are preparing for an audit in 2026, here are the top ten things you must have ready.
Item
Description
1. The Master Schedule
A list of every study that has happened in your lab.
2. Current SOPs
Proof that your team is using the newest versions of your rulebooks.
3. Training Logs
Proof that every person was taught how to do their job before starting.
4. Equipment Records
Logs showing that every scale and fridge is checked regularly.
5. Chain of Custody
A record of who touched a sample at every single minute.
6. Raw Data
Original handwritten notes or machine printouts.
7. QA Reports
Reports from your own internal “referees.”
8. CAPA Plans
Records showing how you found and fixed past mistakes.
9. Computer Validation
Proof that your digital data is secure and cannot be changed.
10. Signatures and Dates
Every page must be signed with no blank spaces or whiteout used.
Final Tip: The Clean Room Rule
An inspector will also look at your physical space. If a lab is cluttered, they will assume the data is also messy. A clean and organized lab tells the inspector that you take quality management seriously. For more help, you can check the FDA Inspection Guide for the latest standards.
Secure Your Future in Science with Metis Consulting Services
When "good enough" no longer passes the test, your organization needs to turn regulatory pressure into a competitive advantage. Contact Metis Consulting Services today to ensure your next breakthrough is backed by an unbreakable foundation of compliance.

Why Your Biotech, Pharmaceutical or Medical Device Organization Needs an Independent Consultant
Navigating the regulatory complexities and laboratory hurdles often hindering the development journey is daunting. This is where independent consultants, such as Metis Consulting Services, play a crucial role in unlocking the full potential of your products and achieving regulatory success.
Written by Li-Anne Rowswell Mufson

In today's complex regulatory landscape, biotech, pharmaceutical, and medical device organizations face numerous challenges in bringing products to market while ensuring compliance with evolving standards. Navigating the regulatory complexities and laboratory hurdles often hindering the development journey is daunting. This is where independent consultants, such as Metis Consulting Services, play a crucial role in unlocking the full potential of your products and achieving regulatory success. Consultants offer fresh eyes that pick up on potential pain points that the team may not see on the inside.
R&D Laboratory
Metis Consulting Services offers comprehensive R&D laboratory support, including but not limited to small molecule development, biologics development services, and cross-functional expertise. With their specialized knowledge and experience, they provide valuable insights and guidance to streamline your development processes and overcome technical challenges. For more, Listen to Queens of Quality Podcast “A Journey through the Pharmaceutical Regulation and Quality Design S2:E7
Audits/Inspection Readiness
Preparing for audits and inspections is critical to maintaining regulatory compliance. Independent consultants assist your organization in conducting thorough audits, ensuring inspection readiness, and implementing corrective actions to address any identified issues, ultimately helping to maintain good standing with regulatory authorities.Ffor more listen to Queens of Quality Podcast A Comprehensive Guide to Audits and Compliance in Pharmaceuticals | S2:E2
Clinical Data Management
Effective clinical data management is essential for demonstrating the safety and efficacy of pharmaceutical and medical device products. Independent consultants offer expertise in clinical data management and ensure that your data is collected, processed, and analyzed in compliance with regulatory requirements. For more listen to Queens of Quality Podcast Unlocking Ethical AI in Life Sciences: Insights with Steve Thompson and Emily Barker PT3
| S2:BONUS 3
Corporate Training
Continuous training and development are vital for keeping your team updated with the latest regulatory standards and industry best practices. Independent consultants provide tailored corporate training programs to enhance your staff's regulatory knowledge and skills, ultimately contributing to improved compliance and operational efficiency. For more listen to: Queens of Quality Podcast A Comprehensive Guide to Audits and Compliance in Pharmaceuticals| S2:E2
Regulatory Strategy Advising
Navigating the complex regulatory landscape requires a strategic approach. Independent consultants advise regulatory strategy, helping your organization develop and execute robust regulatory strategies that align with your business objectives and ensure timely product approvals.For more listen to Queens of Quality Podcast” A Comprehensive Guide to Audits and Compliance in Pharmaceuticals” S2:E2
Pharmacovigilance
Ensuring the safety of pharmaceutical products throughout their lifecycle is a vital concern. Independent consultants support your organization in establishing and maintaining effective pharmacovigilance systems, including adverse event reporting, risk management, and regulatory compliance.
Listen to the Queens of Quality Podcast S1 E5 “Pharmacovigilance”
Quality Management Services
Maintaining high-quality standards across all aspects of your operations is essential for regulatory compliance and product integrity. Independent consultants offer quality management services, including quality system assessments, process improvements, and compliance monitoring, to help you achieve and sustain a culture of quality within your organization. Listen to the Queens of Quality Podcast S2 E8, “Embracing Evolution and Growth in Quality.”
REMS/RMPs
Risk Evaluation and Mitigation Strategies (REMS) and Risk Management Plans (RMPs) are mandatory programs for post-market activities for designated products. REMS and RMPs (in the 2EU) are designed to manage known or potential risks associated with certain products. Independent consultants with expertise in REMS/RMPs guide these strategies' development, implementation, and evaluation to ensure the safe and effective use of pharmaceutical products. Listen to the Queens of Quality Podcast S2 E6, “Revolutionizing the REMS Industry with Sherice Mills.”
Consulting
The expertise and support independent consultants provide can significantly benefit pharmaceutical and medical device organizations, particularly in navigating regulatory challenges, enhancing operational capabilities, and ensuring compliance with the highest standards. Pharmaceutical consulting provides a bridge between laboratory research and patient care. An alliance of scientific rigor and strategic acumen turns therapeutic concepts into accessible treatments. What are the benefits of using pharmaceutical consultants? They guide your organization's journey from bench to bedside in the biotech, pharmaceutical, and medical device industry. Pharmaceutical consultants don the hat of strategists, advisors, and analysts to steer drug developers through the complex maze of bringing a new drug to market. They are the catalysts for innovation, ensuring that groundbreaking treatments see the light of day and reach the right patients at the right time. If your organization is striving for regulatory success and seeking to maximize the potential of its products, engaging an independent consultant such as Metis Consulting Services is an excellent strategic decision with far-reaching benefits, ultimately leading to the primary goal for all involved: enhanced patient outcomes. FDA Guidance on REMS from May 2024 here
Metis Consulting Services offers a complete and customizable suite of services, including but not limited to
1. Audits/ Inspection Readiness-
3. Corporate Training-
4. REMS-RMP
5. Regulatory Strategy Advising-
7. Quality Management Services-
8. R&D Laboratory Services-
9. Ethical AI
With our support, biotech, pharmaceutical, and device manufacturers can confidently navigate the complex landscape of regulatory requirements and industry best practices, ensuring their products' safety, efficacy, and quality.