
ICH Q12: Post-Approval Change Management for Pharmaceutical Product Lifecycle Management
The formalized roll-out of ICH Q12 mechanisms introduces an organized approach to identifying and managing regulatory commitments. This framework allows manufacturers to execute routine modifications under the oversight of their internal Pharmaceutical Quality System (PQS), reducing the burden of prior-approval regulatory filings.
This week in the Guardrail, we break down the practical mechanics of the ICH Q12 framework. Tools like Established Conditions and Post-Approval Change Management Procedures can streamline regulatory paths and protect global supply chains.
By Michael Bronfman
May 29, 2026
The global pharmaceutical regulatory framework is transitioning from a rigid, reactive paradigm to an anticipatory, science and risk-based lifecycle management model. Central to this transformation remains the international implementation of the International Council for Harmonization (ICH) Q12 guideline.
Historically, post-approval changes (PACs) to chemistry, manufacturing, and controls (CMC) required extensive, multi-jurisdictional regulatory reviews. These extended processes frequently delayed the introduction of manufacturing innovations, equipment upgrades, and site transfers.
The formalized roll-out of ICH Q12 mechanisms introduces an organized approach to identifying and managing regulatory commitments. This framework allows manufacturers to execute routine modifications under the oversight of their internal Pharmaceutical Quality System (PQS), reducing the burden of prior-approval regulatory filings.
Evolving Jurisdictional Implementation Boundaries
Global regulatory bodies are adopting the tools and enablers outlined in ICH Q12 at varying paces and within specific product domains.
Health Canada Strategy
Health Canada has introduced updates to its regulatory infrastructure, denoting a step-wise integration of the ICH Q12 framework. The Biologic and Radiopharmaceutical Drugs Directorate (BRDD) updated its Health Canada Guidance on Post-Notice of Compliance Changes Framework to establish the operational boundaries for these tools.
Initial implementation focuses exclusively on Post-Approval Change Management Procedures (PACMPs) for products regulated by the BRDD, including biologics and Schedule C drugs. Under this system, the submission of qualifying PACMPs will be formally accepted following a 90-day transition period ending August 13, 2026.
Notably, Established Conditions (ECs) for all product classes and PACMPs for applications outside the BRDD fall outside the initial scope. Broader integration by the Pharmaceutical Drugs Directorate (PDD) is scheduled for subsequent phases, with particular timelines expected later in the year.
Global Agency Status
The United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) provide precedents for these tools across chemical entities and therapeutic biologics. These agencies accept regulatory submissions containing explicitly defined ECs and PACMPs, provided the manufacturer demonstrates an advanced PQS during routine facility inspections.
The differences in implementation speed underscore the need for multinational pharmaceutical operations to design global change strategies that navigate different regulatory requirements.
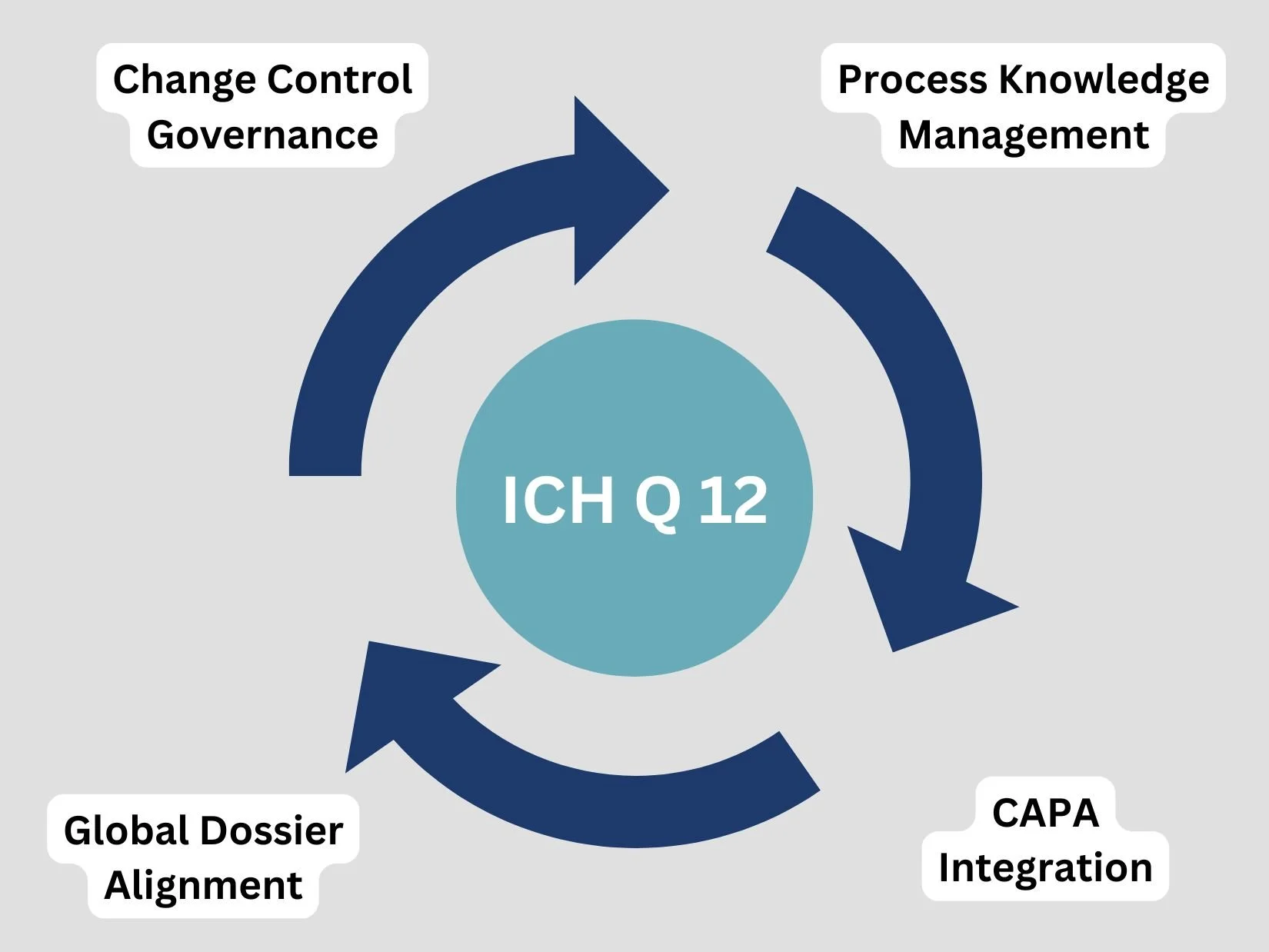
Functional Mechanics of ICH Q12 Regulatory Tools
The practical value of ICH Q12 relies on two interconnected instruments: Established Conditions and Post-Approval Change Management Guidelines. These tools shift the focus of regulatory dossiers from arbitrary data elements to critical-to-quality variables.
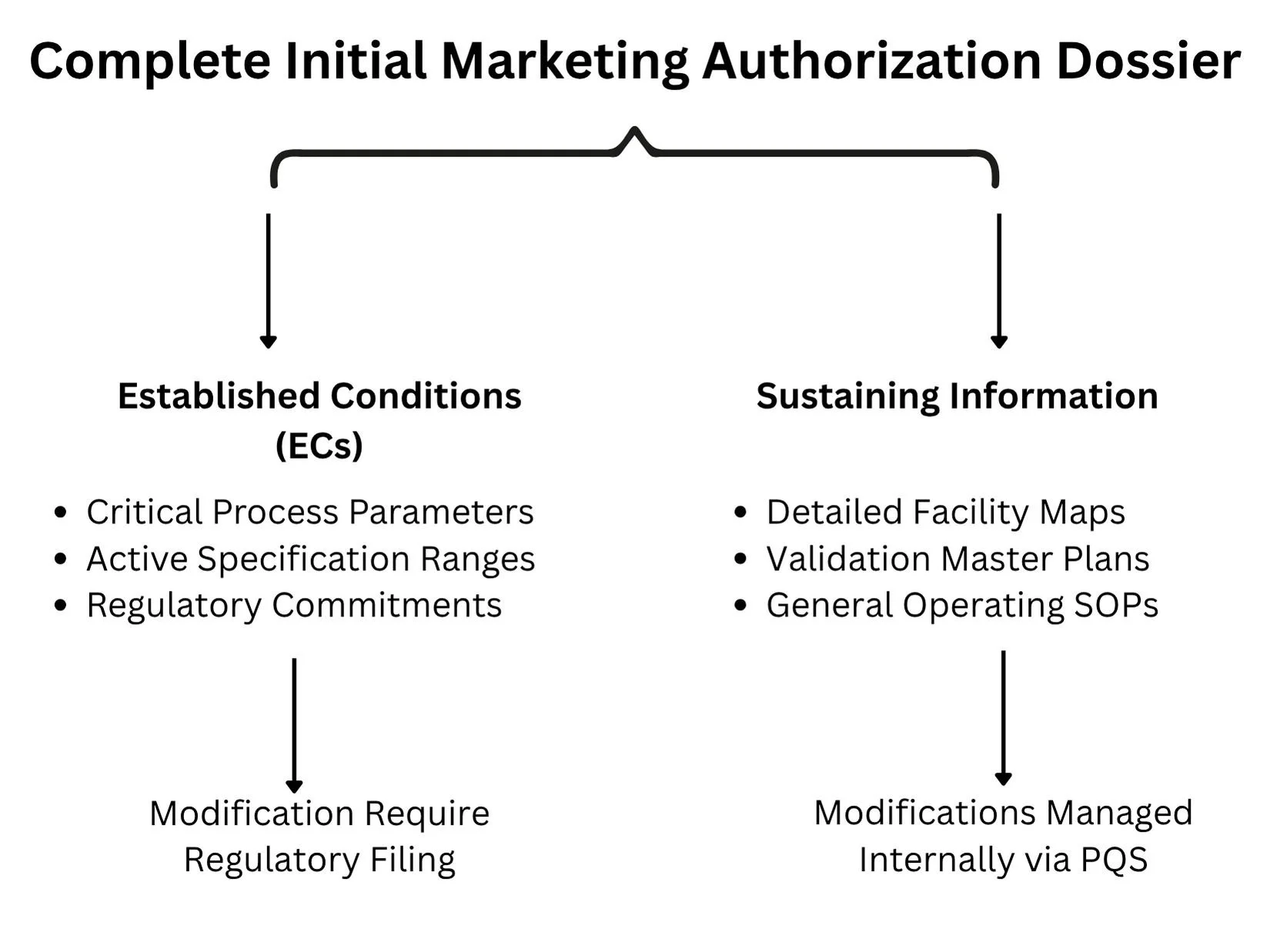
Delineating Established Conditions from Sustaining Information
A longstanding challenge in lifecycle management has been the lack of a clear distinction between legally binding regulatory commitments and purely illustrative descriptive text in the Common Technical Document (CTD). ICH Q12 tackles this by separating information into Established Conditions and Sustaining Information.
Established Conditions (ECs): Defined as the specific elements of a manufacture or control strategy necessary to ensure product safety, identity, strength, purity, or potency. Any modification to an approved EC constitutes a regulatory change that must be reported to the oversight agency. Examples include critical process parameters (CPPs), critical quality attributes (CQAs), acceptance criteria for raw materials, and active operational dimensions of specialized purification columns.
Sustaining Information: Encompasses the underlying science, developmental data, and operational context that supports the designation of ECs. This includes detailed facility blueprints, validation master plans, general operating standard operating procedures (SOPs), and experimental data from early pilot scales. Modifications to sustaining information do not require regulatory notification and are managed entirely via internal site change control protocols.
Post-Approval Change Management Procedures (PACMPs)
A PACMP is a comprehensive, legally binding plan that details a specific manufacturing modification that the sponsor intends to implement throughout the product lifecycle. The protocol explicitly outlines:
The exact nature of the proposed modifications (such as changing an analytical method, upgrading a bioreactor configuration, or transferring an active pharmaceutical ingredient to an alternate facility).
The risk-management strategy is used to evaluate the possible impact of the modification on product quality attributes.
The specific analytical testing, validation matrix, and stability commitments are required to show product comparability.
The predetermined down-regulated reporting category (for example, converting what would typically be a prior-approval Supplement into a post-implementation Notification) if all specified acceptance criteria are met.
By securing prior agency approval for the testing methodology and downgrading logic in the initial PACMP submission, manufacturers can implement modifications quickly once internal testing confirms success.
Structural Requirements of a Mature Pharmaceutical Quality System (PQS)
Sponsors cannot utilize the regulatory flexibilities of ICH Q12 without showing a functional, highly capable PQS that complies with ICH Q10 principles. Regulatory bodies will not grant down-regulated change pathways to facilities lacking robust, data-driven internal quality governance.
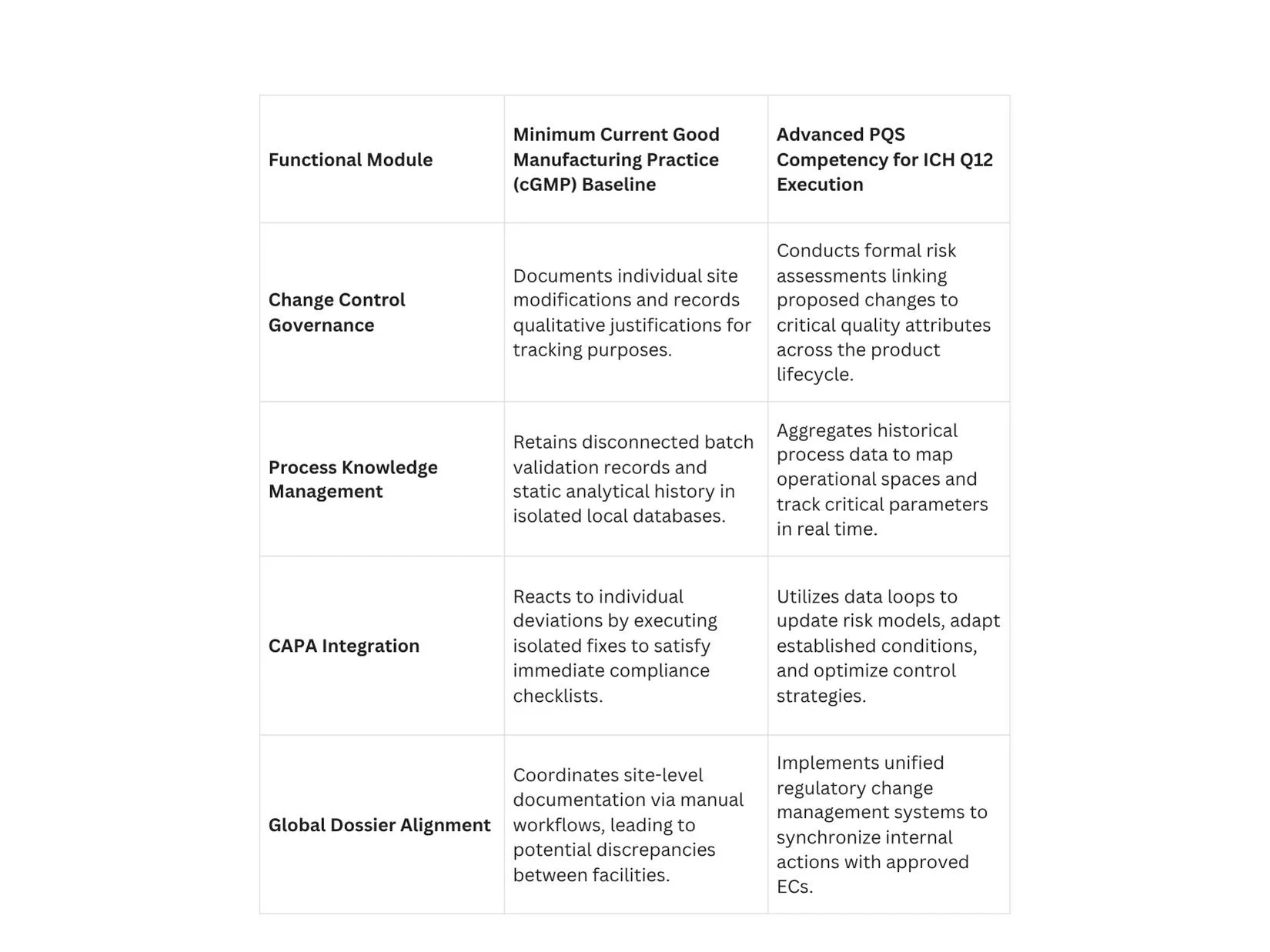
Process Knowledge Management across the Lifecycle
A compliant PQS must operate a continuous knowledge management framework that captures data from early clinical development through commercial manufacturing. Process knowledge should not be stored in isolated paper batch records or disparate local databases.
Instead, it must be aggregated into unified data structures that clearly reflect process parameters, material sources, and environmental variables. This deep process knowledge provides the scientific basis for proposing, justifying, and defending specific boundaries for Established Conditions during regulatory audits.
Regulatory Reporting Categories and Downgrading Strategies
The implementation of ICH Q12 provides a mechanism to modify the default regulatory reporting structures defined by regional laws. The goal is to move low-risk, well-understood adjustments out of prior-approval queues and into post-implementation notification pathways.
The Mechanism of Risk-Based Downgrading
When a manufacturer demonstrates an extensive understanding of a process, they can propose a risk-based categorization strategy for individual ECs. For instance, a process parameter with a broad operating margin and minimal impact on structural attributes can be negotiated from a major reporting tier down to a minor tier.
When this strategy is combined with an approved PACMP, the process efficiency increases significantly. A site transfer for a complex biologic that traditionally required a detailed, prior-approval application can be executed as a post-notice change, provided the verification data satisfies the criteria defined in the protocol.
Introduction of Immediate Notification Pathways
To support such flexibility, modernized regulatory revisions are launching new communication mechanisms. For example, Health Canada introduced a Level III Immediate Notification category within its updated framework. This reporting tier accommodates modifications that have been downgraded from higher risk categories via approved ICH Q12 enablers.
Sponsors utilizing this pathway must notify the agency within 15 days of releasing the modified product to the Canadian market, allowing the regulatory body to maintain oversight without delaying commercial supply lines.
Technical Step-by-Step Implementation Protocol for PACMP Execution
Successfully executing an approved PACMP requires strict adherence to a systematic operational workflow to preserve compliance throughout the product lifecycle.
Phase 1: Protocol Development and Submission
The sponsor prepares a comprehensive PACMP submission within the initial marketing authorization application or via a subsequent formal variation supplement. This document must include a precise description of the future change, the risk mitigation strategy, the analytical methods to be used, and the targeted down-regulated reporting category. The protocol must be reviewed and approved by the target regulatory authority before any subsequent lifecycle modifications can use this pathway.
Phase 2: Internal Facility Execution and Validation
Once the protocol is approved, the manufacturer can initiate the physical change at the designated facility. For example, if transferring production to a new manufacturing line, the site must install the equipment, execute installation and operational qualifications, and run commercial-scale comparison batches.
All analytical data, validation outputs, and stability testing must be conducted exactly as specified in the approved PACMP.
Phase 3: Data Comparison and Acceptance Verification
The quality unit compiles the resulting analytical data and evaluates it against the predetermined acceptance criteria established in the protocol.
If all parameters fall within the approved boundaries, the change is considered successful. If any metric fails to meet the criteria, the protocol becomes invalid for that modification, and the change must revert to the standard prior-approval submission pathway.
Phase 4: Post-Implementation Reporting
Upon verifying compliance, the manufacturer implements the change in commercial production. The sponsor then files the required regulatory notice under the agreed-down-regulated category, such as an immediate notification or an annual report, citing the approved protocol reference number and providing the supporting validation data package.
Commercial and Operational Impact on Global Supply Chains
The transition to an ICH Q12 framework delivers significant strategic and commercial gains that extend beyond basic regulatory compliance.
Mitigating Drug Shortages through Agility
A primary driver of ICH Q12 adoption is the prevention of pharmaceutical supply interruptions and critical drug shortages. In traditional regulatory systems, expanding manufacturing capacity or onboarding an alternative raw-material supplier could take months due to backlogs in prior-approval queues.
By using approved PACMPs and clearly delineated ECs, manufacturers can activate backup manufacturing facilities and alternative material pipelines within days. This nimbleness secures a continuous supply of critical therapeutics to global markets.
Accelerating Ongoing Enhancement and Innovation
The traditional oversight model inadvertently penalized innovation by mandating extensive regulatory filings for minor process improvements. This administrative burden frequently led manufacturers to run outdated processes rather than handle the complex post-approval review landscape.
ICH Q12 removes these barriers, enabling companies to continuously optimize production lines, implement real-time release testing, and deploy advanced process analytical technologies (PAT) under internal PQS controls. This continuous optimization drives lower operating expenses, reduces batch failure rates, and increases overall manufacturing yields.
Conclusion: The Strategic Criticality of Operationalizing ICH Q12
The implementation of ICH Q12 marks a fundamental shift toward an optimized, data-driven approach to pharmaceutical lifecycle management. By implementing tools such as Post-Approval Change Management Procedures and explicitly mapped Established Conditions, manufacturers can significantly reduce regulatory timelines and accelerate facility upgrades.
However, these advanced regulatory flexibilities cannot operate in a vacuum; they require a highly capable, data-driven Pharmaceutical Quality System built on robust knowledge management and risk-based decision-making. As regulatory authorities globally continue to embed these guidelines into their standard oversight frameworks, companies that fail to operationalize these enablers risk a permanent competitive and operational disadvantage.
To review the scientific consensus, emerging clinical data, and peer-reviewed case studies supporting advanced lifecycle management strategies, quality professionals can access Nature.com to ensure their operational systems comply with current international practices.
Don't let rigid regulatory frameworks hold back your manufacturing innovation or compromise your supply chain stability. Contact Metis Consulting Services today.

AI in Regulatory Submissions: Writing for Both Human and Machine Reviewers
This week in the Guardrail, we analyze the dual-audience reality facing modern pharmaceutical compliance. As regulatory agencies integrate automated tools to parse complex submissions, drug sponsors must adapt their documentation strategies to satisfy both algorithmic logic and human expertise.

This week in the Guardrail, we analyze the dual-audience reality facing modern pharmaceutical compliance. As regulatory agencies integrate automated tools to parse complex submissions, drug sponsors must adapt their documentation strategies to satisfy both algorithmic logic and human expertise.
By Michael Bronfman
May 25, 2026
The world of making and approving medicines is going through a massive shift. For decades, pharmaceutical companies wrote drug applications for just one audience: human scientists. Teams of medical doctors, chemists, and statisticians at agencies like the Food and Drug Administration would read thousands of pages of text to decide if a new drug was safe.
Today, that process looks very different. Pharmaceutical companies now use computer algorithms, known as Artificial Intelligence, to run clinical trials and analyze data. At the same time, the regulatory agencies themselves are starting to use computer programs to help read and sort through massive piles of application documents.
This means medical writers and drug sponsors must now write for two very different audiences at the same time. They must write for the human experts who make the final decisions, and they must write for the machine reviewers who scan the text for errors and patterns. If an application is not structured correctly for a machine to read, it could get flagged for inconsistencies before a human expert even looks at it.
To help companies navigate this change, the Food and Drug Administration released official draft guidance about using these advanced computer models in drug development. This document outlines exactly how the agency looks at data generated by computers and how companies should share that information. For more detailed context, you can read the official announcement on the FDA Press Release Page.
The Food and Drug Administration Risk Framework
The official policy from the government makes one thing very clear: not all computer applications are treated equally. The agency uses a risk-based framework to grade how much scrutiny a system needs. This framework is based on two main ideas: model influence and decision consequence.
Model influence means how much the computer output affects the final decision. If a computer makes a final choice on its own, its influence is strong. If a human expert checks the work and can override the computer, its influence is lower. Decision consequence means what could go wrong if the computer makes a mistake. If a computer error harms a patient, the consequences are high. If an error just slows down a factory machine for an hour, the consequence is low.
By looking at these two factors, the government separates computer tools into high-scrutiny systems and low-requirement systems.

High Scrutiny Systems
The highest level of official review is saved for computer systems that directly create evidence for a drug application. These are systems where a mistake could directly hurt a patient or ruin the results of a scientific study.
The government pays closest attention to these five specific areas:
Patient Stratification: Choosing which patients get to be in a clinical trial based on their genetic codes or medical histories.
Dose Optimization: Using mathematical models to calculate exactly how much medicine a patient should take to get better without getting sick from side effects.
Real World Data Analysis: Scanning millions of electronic health records from hospitals to see how a drug performs in everyday life outside of a controlled trial.
Safety Signal Detection: Watching patient data in real time to spot rare and dangerous side effects before they become a widespread public health crisis.
Endpoint Derivation: Using wearable sensors like smartwatches to measure how well a patient is moving or sleeping during a clinical trial.
If a company uses a computer for any of these tasks, it must prove the system is incredibly reliable. They must show how the model was trained, what data it used, and how it avoids bias.
Low-Requirement Systems
On the other side of the coin, some computer uses do not impact patient safety at all. If a company uses a computer tool to format a document, check page numbers, or organize internal administrative tasks, the government does not need to see piles of validation data. These internal operations face proportionally lower requirements because a mistake by the computer will not change the scientific conclusions of the drug trial.
Understanding the Double Audience
Because regulatory agencies are now using advanced software to help manage incoming applications, drug sponsors must realize they are writing for a double audience. The text must satisfy both the human brain and the computer algorithm.
To see how these two audiences read differently, look at this comparison:
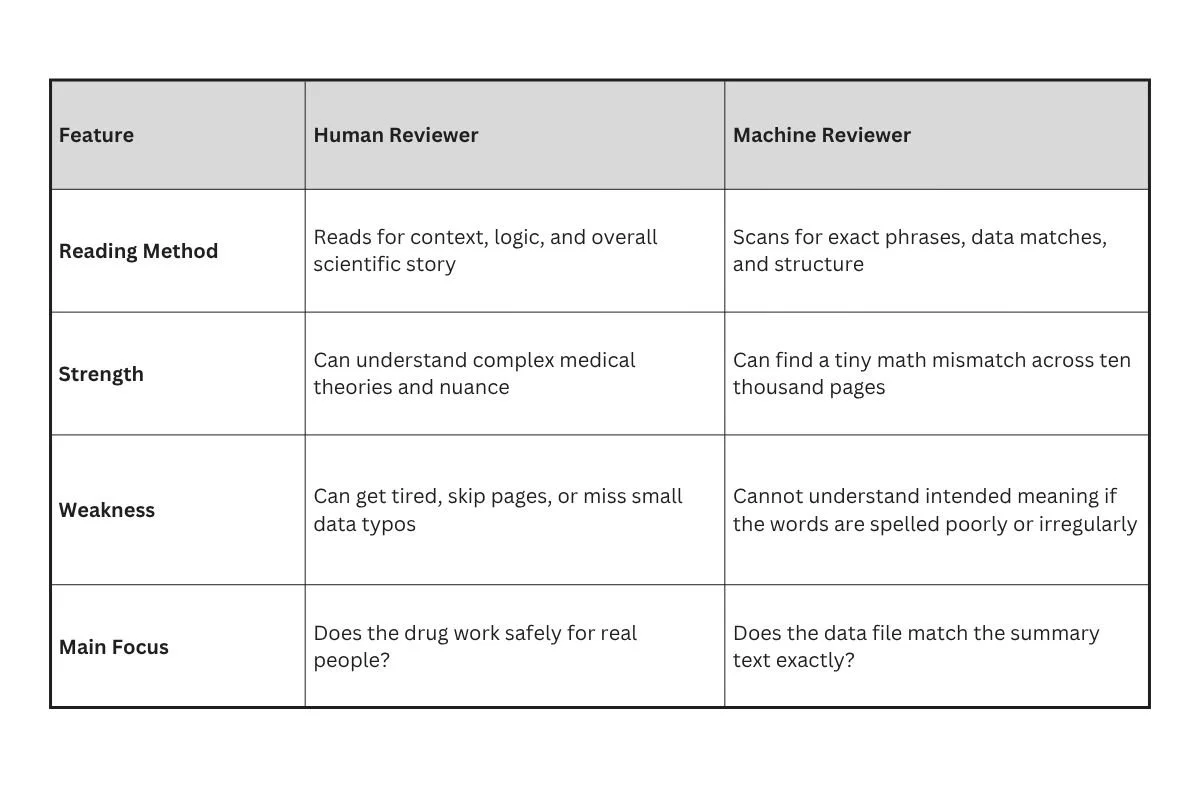
When a human reads a drug application, they want a clear narrative. They want to understand the journey of the drug from the laboratory to the clinic. They care about scientific logic.
A machine reviewer does not care about stories. It treats the document like a database. It looks at the tables, the labels, and the numbers to make sure everything adds up perfectly. If the summary on page five says fifty patients had a headache, but the raw data table on page nine hundred says forty-nine patients had a headache, the machine will flag that instantly. A human might miss that small slip, but a machine never will.
Writing for the Machine Reviewer
Writing for a computer means changing how you present text. Computers like clean organization, predictable patterns, and explicit language. If you write with vague words, the software can get confused and flag your document as a risk.
Structure and Predictability
The best way to help a machine reviewer is to use standard templates. Regulatory documents should follow strict structural rules. Use clear, standardized headings for every section. Do not try to be creative with section titles. If the standard title is Clinical Efficacy, do not change it to How Well the Drug Worked. The computer looks for specific keywords to map the document, and changing those keywords breaks the map.
Data Consistency and Labels
Every data point must look identical throughout the entire file. If you refer to a drug concentration as ten milligrams on one page, do not write it as 10mg on the next page. Choose one format and stick to it.
Also, make sure that every chart and table has clear, descriptive labels that use text instead of scanned images. Machine reviewers read text characters, not picture pixels. If you paste a picture of a table into your document, the computer sees a blank space and misses all the important data inside it.
Front Loading for Clarity
Machines are built to look for core conclusions early. Put your main findings, safety summaries, and essential data points right at the front of your sections. Do not hide your main message under paragraphs of introductory fluff. Front loading your clarity helps the computer categorize your document correctly on its very first pass.
Writing for the Human Reviewer
While you must make your document easy for a computer to analyze, you cannot forget the human being who must sign the final approval paper. Humans need context, clear explanations, and a believable scientific argument.
Explaining the Why
A machine can show that a number changed, but only a human can explain why it changed. If a clinical trial had a sudden drop in patient attendance during month four, a machine might flag it as a data error.
The human writer needs to explain the context:
"Patient attendance dropped in month four due to a historic blizzard that closed three major clinical trial sites for two weeks, but patients resumed their regular visits as soon as the roads cleared."
This explanation satisfies the human reviewer and prevents them from rejecting the data.
Keeping the Story Alive
A good regulatory submission tells a story of safety and success. The human writer must connect the dots between different pieces of research. Show how the animal studies predicted the human results, and show how the human results match the goals of the project. Use active, plain verbs to explain what the scientists did. Avoid overly dense language that puts the reader to sleep. A tired reviewer is a frustrated reviewer.
Conducting an Internal Review
Before you click the submit button to send your drug application to the government, your team should perform a complete internal review. This means testing your document against your own software tools to see what a machine reviewer will find.
Step One: The Automated Consistency Check
Run your completed document through text-matching software. This program should look for every number, percent, and statistical value to make sure they match perfectly across all chapters. If the software finds a conflict, fix it immediately. You want to find these errors yourself rather than letting the government find them first.
Step Two: The Structure Audit
Verify that every hyperlink works and leads to the correct appendix. Check that your document map functions properly and that all headings match the standard table of contents. If a machine cannot navigate your document links, it may automatically reject the file.
Step Three: The Human Readability Pass
Have a scientist who did not write the document read it for flow and clarity. Ask them if the arguments make sense and if the explanations are easy to find. This step ensures that once your document passes the computer gates, it will please the human experts.
The Path Forward for Drug Developers
The use of computer intelligence in regulatory submissions is not a temporary trend. It is the permanent future of medicine. Drug companies that learn how to write for both humans and machines will get their medicines approved much faster. Those who stick to old ways of writing will face constant delays, data flags, and rejection notices.
To keep up with these changes, companies should train their medical writers in basic data science principles. Writers do not need to learn how to code, but they do need to understand how computers read and sort information. By focusing on predictability, exact data matches, and clear summaries, you can create a document that satisfies the cold logic of a machine and the deep wisdom of a human scientist.
To learn more about how the government views these new digital tools, you can review the comprehensive resources provided by theFDA Artificial Intelligence Development Page. Staying informed about these official updates is the best way to ensure your future submissions are successful.

Writing for Human and AI Reviewers: The New Way to File
The rise of automated FDA AI reviewers means mastering the balance between machine-readable data and clear medical storytelling. It is no longer optional—it is the key to avoiding costly filing delays.

The rise of automated FDA AI reviewers means mastering the balance between machine-readable data and clear medical storytelling. It is no longer optional—it is the key to avoiding costly filing delays.
By Michael Bronfman
May 11, 2026
The world of medicine is changing fast. For decades, pharmaceutical companies followed a simple path: run a study, write a report, and send it to the Food and Drug Administration (FDA). The “audience” was always a group of human scientists. But in 2026, the rules have shifted. Today, when a company submits a new drug application, the first “eyes” on the document might not be human at all.
Smart software and advanced data tools now help regulators look through thousands of pages in seconds. This means that if you are writing a regulatory submission, you are no longer just writing for a doctor or a chemist. You are writing for a machine, too. This double audience requires a whole new way of thinking about how we present science.
Why This Matters Now
The FDA recently released new rules about how companies can use smart technology in their filings. They made a big distinction between “low risk” and “high risk” uses. This matters because it tells companies where they need to be the most careful and spend the most time.
High Risk: If a computer program is used to select which patients receive a drug, determine the dose, or identify safety signals, the FDA looks at it very closely. This is because these tasks directly impact whether a drug is safe for people. This is the area of high scrutiny.
Low Risk: If the technology is just helping with internal office work, scheduling meetings, or organizing files, the requirements are much lighter.
Because of these new rules, companies have to change how they communicate. They have to be clearer and more organized than ever before. If a machine cannot understand your report, it might flag it as a mistake, even if the science is perfect. A flag from a machine can lead to months of delays, costing companies millions of dollars and keeping medicine away from patients who need it.
Writing for the Machine: What Does It Mean?
Machines do not read as we do. They do not look for beautiful prose or clever metaphors. They do not get impressed by fancy vocabulary. Instead, they look for patterns, data points, and absolute consistency. To get a submission through a machine review without any red flags, writers must use a front-loading strategy.
Front Loading Clarity
“Front loading” means putting the most important information at the very beginning of every section. Instead of building up to a conclusion like a mystery novel, you state the conclusion first.
Old way: After reviewing 500 patients over 6 months and checking their blood pressure daily, we found that the drug worked.
New way: The drug reduced blood pressure by 15% in 500 patients. This conclusion is based on a six-month study where…
This helps the machine categorize the information instantly. It creates a “map” for the software to follow.
Avoiding Inconsistencies
One of the biggest reasons a filing gets flagged today is a data mismatch. Imagine you say a drug is 90% effective on page 10, but a table on page 400 says 89.9%. A human might realize it is simply a matter of rounding up and continuing reading. A machine sees a red flag and stops.
To prevent this, companies are now doing AI readiness reviews. This is a step where the company runs its own software on the document before sending it to the government. They look for the same things the FDA’s machines will look for:
Terminology: Using the exact same word for a concept every single time. Do not call it “the medicine” in one spot and “the compound” in another if you want the machine to track it easily.
Cross References: Making sure every link to a chart or table actually works and points to the precise data.
Structure: Following the eCTD format (electronic Common Technical Document) perfectly, so the software knows where to look for information.
The Human Factor: Keeping the Science Real
Even though machines are doing the heavy lifting, humans still make the final decision. A doctor at the FDA still needs to trust that the drug works. This creates a double challenge. You have to be technical enough for a computer but clear enough for a person.
The Problem with “Robot Speak”
Sometimes, when people try to make things easy for computers, the writing becomes stiff and hard to follow. This is a mistake. If a human reviewer gets confused ot just bored, they may start to doubt the work. The best regulatory writing today uses plain language principles.
Short Sentences: Long, winding sentences may confuse both people and software. Aim for 20 words or fewer.
Active Voice: Saying “The study showed…” instead of “It was shown by the study…” makes the facts stand out and defines who is responsible for the action.
Bullet Points: Lists are easy for machines to scan and for busy human reviewers to read quickly during a long workday.
High Scrutiny Areas: Where Accuracy Counts Most
The FDA Guidance for Industry focuses heavily on a few specific areas. If your submission uses advanced tech for these, expect the highest level of checking:
1. Patient Stratification
This is a fancy way of saying that patients are being sorted into groups. If a computer picks which patients will benefit most from a drug based on their DNA or history, the FDA wants to know exactly why. You cannot just say “the computer said so.” You have to explain the logic in a way a human can verify.
2. Dose Optimization
Finding the right amount of medicine to give someone is a science. If you use a machine to find that “perfect dose,” you must prove the machine isn’t making a mistake that could hurt someone. This requires showing the “math” behind the machine’s decision.
3. Real World Data Analysis
Sometimes companies analyze health records from millions of people to see how a drug works in the real world. This is a mountain of data. Machines are great at this, but they can also find patterns that don’t actually exist (called “noise”). Your report must explain how you ensured the data was clean and the patterns were genuine.
4. Safety Signal Detection
This is about finding side effects. If a machine is the first thing to “notice” a side effect in a clinical trial, the documentation must show how that information was passed to human doctors for a final check. The human must always be in the loop.
The Importance of Pre-Submission Checks
In the old days, a team would proofread a document for typos and then send it off. In 2026, that is not enough. The “Internal AI Readiness Review” is now a required step for any serious pharma company.
This process involves using tools to “stress test” the document. For example, the team asks:
“Can a computer find the primary endpoint in less than one second?”
“Are there any hidden characters or weird formatting that will break the FDA’s software?”
According to Clinical Leader experts, companies that skip this step often face “Refusal to File” letters. This means the FDA will not even look at the science because the document itself is too messy for their tools to handle.
The Role of the Medical Writer in 2026
The job of a medical writer has changed. It is no longer just about writing; it is about information architecture. A writer today must understand how data flows from the lab into a table and convey that with a paragraph.
They act as a bridge. On one side, they have the data scientists who talk in code and numbers. On the other side, they have the regulators who want to ensure public safety. The writer must translate complex data into a structured format that satisfies both software scanners and human doctors. This requires a deep understanding of the eCTD structure and the ability to write with mathematical precision.
How to Prepare: A Practical Checklist
If you are working on a pharma team, you cannot wait until the last minute to think about these things. Preparation starts months before the “submit” button is pushed.

Ethics and Transparency: The “Explainable” Requirement
One thing a machine cannot do is be ethical. It cannot think about the spirit of the law or the “heart” of a patient. That is why transparency is the biggest buzzword in 2026.
When you use a machine to help write or analyze a filing, you must be honest about it. You must show the pathway the machine followed to reach its answer. This is often called Explainable AI. If a regulator can see the steps, they can trust the result. If the process is obscured, a “black box” in which no one knows how the answer was derived, the FDA will likely reject it.
Bridging the Gap
Writing for both human and machine reviewers is a new skill, but it is one that every professional in the pharmaceutical industry needs to learn. By focusing on structure, consistency, and clear language, companies can get life-saving drugs to patients faster.
The goal should not be to let the machines take over the process. Instead, the goal is to use the machines to make our work more accurate and organized. This allows FDA staff to spend less time looking for errors and more time examining the science. When we write for both audiences, everyone wins—especially the patients waiting for new treatments.
For more information on the technical side of these filings and to stay updated on new research, you can explore the Wiley Online Library.
Key Takeaways for understanding
Machines are now helping regulators read drug reports. Because of this, we have to write in a way that doesn’t confuse the software.
Consistency is king. If you use different words for the same thing or have small math errors, the machine will flag it as a big problem.
The FDA cares most about “High Risk” tasks. If a computer is used to determine a patient’s dose or identify safety issues, the rules are much stricter.
Clear writing helps humans and computers. Short sentences, bullet points, and putting the main point first (front loading) make the report better for everyone.
Always do a “practice run.” Companies now use their own software to check their reports for mistakes before sending them to the government.
Don’t let a technical red flag stand between your breakthrough and the patients who need it most. Contact Metis Consulting Services today to ensure your next submission is AI-ready, human-approved, and built for success.

How the FDA is speeding up Psychedelic Therapies with National Priority Vouchers
How the FDA is speeding up Psychedelic Therapies with National Priority Vouchers

By Michael Bronfman
May 4, 2026
This week in the Guardrail, How groundbreaking FDA policy shifts are accelerating the path to market for psychedelic-assisted therapies. New incentive structures for mental health may finally bridge the gap between clinical innovation and patient access.
Mental health care is undergoing a dramatic transformation. For decades, the standard treatment for depression and anxiety relied on the same types of pills—some effective, many not. Now, government and scientific attention are rapidly shifting to "psychedelic" medicines like psilocybin (from magic mushrooms) and methylone, which provide prospects for those left behind by previous treatments.
Just this month, in April 2026, the Food and Drug Administration (FDA) made a historic move. They awarded special rewards, called National Priority Vouchers, to three groups working on these new treatments. These vouchers are like a "fast pass" at a theme park. They allow a company to jump to the front of the line when the FDA checks its new drug. This could mean life-saving medicine reaches patients months or even years earlier than usual.
What is a National Priority Voucher?
To understand why this is a big deal, we have to look at how drugs get approved. Normally, it takes the FDA a long time to review all the data from a clinical trial. They want to make sure a drug is safe and that it actually works. This process can take ten months or more.
A Priority Review Voucher (PRV) changes the rules. It tells the FDA they must complete their review in about 6 months, rather than 10. Recently, a new type of voucher, the Commissioner’s National Priority Voucher (CNPV), was created. These were specifically designed to help fight the mental health crisis.
In April 2026, President Trump signed an executive order to help veterans and other people struggling with mental illness. He told the FDA to use these vouchers to speed up the review process for drugs with "breakthrough" potential.
Let's look at who is driving this change and receiving these important vouchers.
Three major groups received these special vouchers on April 24, 2026:
Compass Pathways: They are testing a synthetic version of psilocybin called COMP360 for people who have "treatment-resistant depression." This means the patients have tried at least two other pills that did not work.
Usona Institute: A non-profit group. They are using psilocybin to treat "major depressive disorder," which is a severe form of sadness that makes it hard to live a normal life.
Transcend Therapeutics (now part of Otsuka): They are working with a drug called methylone to treat Post Traumatic Stress Disorder (PTSD). This is especially important for military veterans who have seen combat.
Why These Drugs Are Called "Breakthroughs"
The FDA does not give these vouchers to just any drug. A medicine must first earn a Breakthrough Therapy Designation. This title is given when early tests show that a drug might be much better than what we already have.
Psychedelic therapies are different because they aren't simply a pill you take every morning. Usually, a patient takes the medicine once or twice in a doctor's office while a trained therapist guides them through the experience.
Breaking the Cycle of Depression
For people with "treatment-resistant depression," life can feel like being shut in a dark room with no door. Standard drugs often simply dull the pain. Researchers compare psychedelics to a "reset button" for the brain, helping it build new connections.
In recent Phase 3 trials—the final step before a drug is sold—Compass Pathways found that some patients felt better within a single day. Those effects lasted for six months for many people. This is a huge leap forward compared to older drugs that take weeks to start working.
The Big Goal: Helping Our Veterans
One of the main reasons the government is pushing so hard for these vouchers is to help veterans. Many soldiers come home with PTSD. They might have nightmares, feel angry, or feel very alone. Sadly, current treatments do not help everyone, and suicide rates among veterans are very high.
The government is now using every tool available to fix this. By giving vouchers to companies like Transcend and Usona, they are saying that mental health is a national priority.
The new rules also talk about the Right to Try Act. This law allows patients who are very sick to try experimental drugs before the FDA fully approves them. This is being expanded to include psychedelic compounds so that people who have no other options can get help now.
How the Vouchers Work Behind the Scenes
You might wonder why a company needs a "voucher" to go faster. Isn't every drug important? The truth is that the FDA is very busy. They have thousands of applications to read.
When a company uses a voucher, they also have to pay a large fee. In 2025, that fee was about $2.5 million. This money helps the FDA hire more staff so they can review papers faster without slowing down other important drugs, such as those for cancer or heart disease.
One common question is about whether these vouchers themselves can be sold, as has happened in other programs.
In the past, certain types of vouchers could be resold—sometimes for over $100 million. Typically, a small company would be awarded a voucher for developing a rare disease drug and then sell the voucher to a large company, using the funds to support further research.
However, the new Commissioner’s National Priority Vouchers (CNPVs), given specifically for psychedelic treatments, are different. Reports indicate that these particular vouchers cannot be sold; only the company that earned them can use them. This rule makes sure that the experts who did the hard work are the ones who bring the medicine to market and prevents the resale that happened with other FDA voucher types in the past.
Going forward, it’s important to consider the following steps for these therapies and the wider mental health environment.
Even with a fast pass voucher, the work is not over. The companies still have to finish their big Phase 3 studies. They have to prove that the drug is safe over a long period of time.
The FDA is also expected to release new "final guidance" very soon. This will be a rulebook for how all future psychedelic drugs should be tested. It will cover things like:
How many therapists need to be in the room?
How to keep the patients safe during the "trip."
How to measure whether the drug is actually improving the patient's life.
A Timeline for Change
If everything goes well, we might see the first fully approved psychedelic medicine by the end of 2026 or early 2027. Because of the vouchers, that date moved up by at least four months. In the world of mental health, four months can save thousands of lives.
Common Questions About the New Vouchers
Are these drugs legal now?
No. These drugs are still "investigational." That means they are only legal to use in special research studies or through the "Right to Try" program for very sick people. They are not yet available at a local pharmacy.
Does a voucher guarantee approval?
No. A voucher merely guarantees a faster review. The FDA can still say "no" if it thinks the drug is unsafe or the data are not good enough. For example, a group called Lykos tried to get an MDMA drug approved for PTSD, but the FDA said no because they needed more data. The voucher just speeds up the "yes" or "no" response.
Why is this happening in 2026?
The mental health crisis has reached a point where the government has decided to take bold action. By using executive orders and creating new voucher programs, leaders are trying to solve the problem faster than the old system allowed.
A Future of Promise
The use of National Priority Vouchers for psychedelic therapies is more than merely a boring business move. It is a signal that our society is ready to think differently about mental health. We are moving away from daily pills that only mask symptoms and moving toward treatments that might actually heal the brain.
By giving these "fast passes" to scientists, we are giving precedence to the millions of people—including our brave veterans—who have been waiting for a breakthrough. The road is still long, and there are many rules to follow, but for the first time in decades, the finish line is in sight.
The year 2026 will likely be remembered as the year the "psychedelic revolution" finally got the go-ahead from the highest levels of government. It is an exciting time for science, and an even more hopeful time for patients.
Important Links to Follow the News
If you want to keep track of these changes, here are some active websites where you can find the latest updates:
FDA Press Announcements https://www.fda.gov/news-events/fda-newsroom/press-announcements – This is where the government officially announces new vouchers and drug approvals.
ClinicalTrials.gov – You can search for "psilocybin" or "PTSD" to see what studies are happening near you.
Psychedelic Alpha – A news site that tracks the business and law side of these new medicines.
Usona Institute – The website for the non-profit group that just received a national priority voucher.
Compass Pathways – Information on the Phase 3 trials for treatment-resistant depression.
Regulatory changes for psychedelic medicine are moving faster than ever, and navigating the complexities of FDA vouchers and breakthrough designations requires expert precision. Ensure your company is positioned at the front of the line—contact Metis Consulting Services today. The strategic guidance you need to bring life-changing healing to those who need it.

GLP and GCP: How the FDA and EPA Watch Over Science
Two of the most important sets of rules are called GLP and GCP. In 2026, the two main government agencies responsible for these rules, the Food and Drug Administration (FDA) and the Environmental Protection Agency (EPA), are working together more than ever before. Re: FDA & EPA Oversight.

This week in the Guardrail…
We explore how federal oversight is shifting standards in 2026, making rigorous data integrity the new baseline for every laboratory and clinic.
By Michael Bronfman
The world of making new medicines and chemicals is a very busy place. Every day, scientists are working in labs and clinics to find the next big cure or a safer way to clean our homes. Because these products can affect our health and the planet, the government has very strict rules to ensure everything is done correctly. Two of the most important sets of rules are called GLP and GCP. In 2026, the two main government agencies responsible for these rules, the Food and Drug Administration (FDA) and the Environmental Protection Agency (EPA), are working together more than ever before. This “shaking out” of the rules is changing how companies operate.
What Do These Letters Mean
To understand the future of science, you first have to know what these abbreviations stand for. They are like a specialized language for safety and honesty.
GLP stands for Good Laboratory Practice. These rules are for the early stages of research. This is the work that happens in a lab with test tubes, plants, or animals before a human ever touches the product. The Environmental Protection Agency uses these rules to ensure that a new pesticide or powerful cleaner will not harm the environment or the people using it.
GCP stands for Good Clinical Practice. These rules start when the research moves into a clinic and involves human volunteers. The Food and Drug Administration uses GCP to ensure that participants in medical studies are safe and that the results are truthful.
The FDA and the EPA Joining Forces
In the past, these two agencies mostly stayed in their own separate worlds. If a company were making a heart medicine, they would talk to the FDA. If they were making a new bug spray, they would talk to the EPA. But today, many new products fall into both categories. For example, a special soap that kills germs on your skin might be considered both a medicine and a chemical.
Because of this, the FDA and EPA are now sharing their notes. If an EPA inspector finds that a lab is messy or that the scientists are not recording their results correctly, they report it to the FDA. This means companies can no longer be “half good.” They have to follow the rules perfectly for both agencies. This coordination ensures that, no matter what kind of product is being made, the public is protected by the highest standards.Official Federal Register : How these agencies work together
Why Honesty Is the Only Policy
The main goal of both GLP and GCP is to ensure data integrity. Data is just a fancy word for the information and results gathered during an experiment. If a scientist says that a drug worked on ten people, the government wants to see the actual signatures and blood test results to prove it.
If a company is caught lying about its data, it can be fined millions of dollars. They might even be banned from ever making products again. This is why risk management is so important during the middle stages of a study. If a company finds a minor mistake in its lab records, it needs to fix it immediately. Waiting until later to “clean up” the paperwork is a huge risk that can lead to a total failure when the FDA or EPA comes to visit.
Protecting the People in the Studies
While GLP protects the science in the lab, GCP protects the people in the clinics. Every person who joins a clinical trial is a volunteer. They are doing something brave to help others. GCP rules ensure that these volunteers are treated with respect.
Under these rules, every volunteer must sign a form acknowledging the risks. This is called informed consent. The doctors must also closely monitor the volunteers for any side effects. If a patient experiences a headache or dizziness, it must be recorded in the official files. TheNational Institutes of Health provides extensive information on how these rules help keep people safe in medical research.
The Quality Control Revolution
In 2026, many companies are hiring specialized teams just to ensure quality. These teams are like the “referees” of science. They do not do the experiments themselves. Instead, they monitor the other scientists to ensure they follow all GLP and GCP rules.
They check that lab machines are properly calibrated. They check to ensure that every signature on a form is genuine and dated correctly. This might sound like a lot of extra work, but it saves the company from failing an inspection. When a company has a high “quality score,” the FDA and EPA can trust their results much more easily. Organizations like theSociety of Quality Assurance help train these specialized workers to stay up to date on the latest rules.
Using Technology to Stay Safe
Technology is making it easier for the FDA and EPA to do their jobs. In the old days, inspectors had to look through thousands of paper files. Now, most of the data is digital. This allows the government to look at the data in real time.
If a lab in California runs a test, an official in Washington, D.C., can see the results almost instantly. This helps catch mistakes before they become big problems. It also makes it harder for anyone to change their results later to make a drug look better than it really is. This transparency is a big part of why the “shake out” between the two agencies is happening so fast. Digital tools make it impossible to hide in the shadows.
The Global Impact of These Rules
Australia, Europe, and the United States all have their own versions of these rules. However, they are all starting to look very similar. This is good news for the public. It means that a drug tested in Australia can be sold in America as long as it follows the same high standards of GLP and GCP.
When countries agree on the rules, medicines can travel around the world much faster. This helps people in every country get the help they need without waiting years for additional testing. TheWorld Health Organization works to help all countries follow these same high standards for health and safety.
Education Is the Key
For these rules to work, every person in the pharmaceutical industry needs to be educated. It is not just the boss's job. Even the person cleaning the lab or the nurse at the clinic needs to understand why the rules matter.
Education helps people understand that following the rules is about more than just avoiding a fine. It is about making sure that the medicine your grandmother takes, or the soap you use on your children, is 100 percent safe. When everyone knows the “why” behind the rules, they are much more likely to follow them correctly every single day.
Risk Management and Quality Systems
Modern pharmaceutical companies use a Quality Management System (QMS) to track everything. A QMS is like a giant digital brain that stores all the company's rules and records. It helps with risk handling by flagging errors the moment they happen.
In 2026, risk management strategies are no longer just about fixing problems. They are about predicting them. By using clinical data management tools, a biotech firm can detect whether a machine is starting to wear out or whether a lab is experiencing too many small errors. This type of risk management planning helps prevent major disasters that lead to FDA warning letters.
The Role of REMS in Safety
Another way the FDA keeps people safe is through REMS, which stands for Risk Evaluation and Mitigation Strategies. These are extra safety programs for drugs that might be dangerous if not used exactly right. For example, some medicines require a patient to get a blood test every month. TheFDA REMS website tracks these programs to ensure drug companies are doing their part to manage risk.
Final Thoughts on the Future of Quality
The “shaking out” of rules between the FDA and the EPA is a positive step for everyone. It means that science is becoming more open and more honest. Companies are learning that they cannot take shortcuts during any stage of research.
By following the rules of GLP and GCP, we ensure that the future of medicine is bright. We can trust the products we buy because we know the government is watching and the scientists are being careful. Quality is not just a set of letters; it is a promise to the public that their safety is the most important thing of all. Don’t wait for a problem to appear before you start being careful. Start with quality on day one and the rest of the journey will be much smoother for everyone.
Frequently Asked Questions: GLP, GCP, and Regulatory Compliance
What is the main difference between GLP and GCP?The biggest difference is when they are used. GLP is for the preclinical stage, which is work done in a lab on animals or cells. GCP is for the clinical stage involving human volunteers.
Why does the EPA care about laboratory rules?The EPA ensures that chemicals like weed killers do not pollute our water. They use GLP to ensure companies are honest about chemical safety. You can find guidelines on theEPA Compliance Monitoring page.
Can a company fail a trial if they follow the science but miss the paperwork?Yes. In the eyes of the FDA, if a test was not documented correctly, it never happened. “Clean data” is just as important as the medicine itself.
What happens during an FDA or EPA inspection?Inspectors check original notebooks, computer logs, and even equipment logs. They want to see a clear trail from the study's start to the final report.
How does Risk Mitigation help with these rules?It means finding small mistakes before the government does. Fixing a mistake early in a study costs much less than fixing it later, when thousands of people are involved.
Where can I stay updated on these changing rules in 2026?The best place to watch for updates is theFDA Voice blog. This site tracks how the agencies are joining their rules for digital data. https://www.fda.gov/news-events/fda-newsroom/fda-voices.
The Pre-Inspection Compliance Checklist
When an inspector arrives, they look for a “culture of quality.” If you are preparing for an audit in 2026, here are the top ten things you must have ready.
Item
Description
1. The Master Schedule
A list of every study that has happened in your lab.
2. Current SOPs
Proof that your team is using the newest versions of your rulebooks.
3. Training Logs
Proof that every person was taught how to do their job before starting.
4. Equipment Records
Logs showing that every scale and fridge is checked regularly.
5. Chain of Custody
A record of who touched a sample at every single minute.
6. Raw Data
Original handwritten notes or machine printouts.
7. QA Reports
Reports from your own internal “referees.”
8. CAPA Plans
Records showing how you found and fixed past mistakes.
9. Computer Validation
Proof that your digital data is secure and cannot be changed.
10. Signatures and Dates
Every page must be signed with no blank spaces or whiteout used.
Final Tip: The Clean Room Rule
An inspector will also look at your physical space. If a lab is cluttered, they will assume the data is also messy. A clean and organized lab tells the inspector that you take quality management seriously. For more help, you can check the FDA Inspection Guide for the latest standards.
Secure Your Future in Science with Metis Consulting Services
When "good enough" no longer passes the test, your organization needs to turn regulatory pressure into a competitive advantage. Contact Metis Consulting Services today to ensure your next breakthrough is backed by an unbreakable foundation of compliance.