
Moving Beyond the Sandbox: How to Build Inspection-Ready AI in Regulated Life Sciences
For years, AI was just a tool for simple tasks like drafting emails. Those days of casual use are over. Regulators are tightening expectations, demanding strict AI governance, perfect traceability, and full integration with quality compliance systems. These rules are no longer optional best practices.
As global regulators rapidly tighten enforcement on pharmaceutical companies, this week we look at the high-stakes gap between flashy AI pilots and the rigorous, audit-ready validation required in the life sciences sector. Manufacturers must have systems in place for bulletproof governance to survive their next inspection.
By Michael Bronfman
June 30, 2026
Many pharmaceutical companies live in a frustrating middle ground. You can see it in boardrooms and IT departments everywhere. Teams enthusiastically say, “We are experimenting with artificial intelligence!” Yet when asked whether that same technology is ready for an official regulatory inspection, the room falls silent.
The gap between a cool pilot project and a fully validated, inspection-ready system is where most life sciences companies currently find themselves.
For years, teams treated artificial intelligence as a collection of non-product tools used for simple tasks. It might have been used to summarize long documents or draft email templates. But the days of casual experimentation are officially over. Regulatory bodies are tightening their expectations. They are demanding strict AI governance, perfect traceability, and complete integration with quality compliance systems. They are no longer leaving these rules to optional best practices.
Artificial intelligence systems inform labeling, product performance claims, drug dosing, patient safety, or quality decisions; they face a tough reality. The entire solution must fulfill rigorous quality, validation, and lifecycle controls. Generic pilots fail to scale because they lack the foundation required to survive a regulatory audit. Life sciences organizations must shift to purpose-built artificial intelligence that incorporates strict governance controls, unalterable audit trails, and validated results to succeed.
The Shift in Regulatory Reality
Why do generic pilots fail?
To understand this, look at how global authorities view technology. The Food and Drug Administration released major updates that signal a strong enforcement posture for advanced software used in regulated spaces. The agency treats high-risk artificial intelligence with the same seriousness as physical medical devices or critical manufacturing tools.
Traditional software validation worked well for static systems. In the past, computer systems validation followed a predictable path. A developer wrote code, a quality team tested that it did exactly what it was supposed to, and the software never changed unless an engineer manually updated it.
Artificial intelligence contradicts this old way of thinking. Advanced models are dynamic. They are built to learn, observe patterns, and develop over time. Because these systems can evolve based on the information they process, standard testing methods are inadequate. Software cannot be tested once and assumed to behave exactly the same way a year from now.
Regulators are fully aware of this challenge. They are looking closely at:
Design Controls: How the model was built, chosen, and structured.
Model Validation: Proof that the mathematical formulas produce accurate, repeatable results.
Data Authenticity: Complete certainty that the information feeding the model is clean and unaltered.
Risk Management: Clear plans for handling unexpected errors.
If an inspector walks into your facility today and sees an advanced tool helping your quality team make decisions, they will ask tough questions. They will want to know how you verify the output. They will want to see how you track changes in the system. If your only answer is that a vendor told you the tool works, you are facing a major compliance risk.
Why Generic AI Pilots Fail to Scale
It is incredibly easy to build a successful pilot project. A small team can upload historical quality data into a popular, generic large language model. Within an afternoon, the tool can review past records and suggest draft standard operating procedures or summarize corrective and preventive action reports. The team celebrates, declares the project a success, and plans to roll it out to the whole company.
Then, they meet the quality assurance department.
Generic artificial intelligence applications are built for mass productivity, not the high-stakes world of life sciences. When you try to push a basic pilot into a Good x Practice environment, the system usually falls apart for several reasons.
The Opaque Decision Process
Generic models operate like a black box. A user submits a question, and the tool provides an answer, but no one can trace the exact path the software took to reach that conclusion. In a regulated environment, an untraceable answer is a non-compliance finding waiting to happen. If you cannot prove how your software reached a conclusion about a batch failure or a clinical trial data point, you cannot use that conclusion.Missing Explicit Intended Use
Validation cannot be generic. You cannot validate an advanced tool for general office work and then use it to triage quality investigations. Every application must have a clearly defined intended use statement. This statement must outline the exact process the tool supports, who the users are, what source systems feed it data, and how the output impacts human health or product quality. Generic tools are not built to restrict themselves to a single, tightly controlled workflow.Model Drift and Information Degradation
When an advanced model interacts with new data, its internal weights can shift. Over time, the system's accuracy can degrade or change, a phenomenon termed as model drift. Generic applications do not include built-in alerts that notify you when the software becomes less accurate. Missing continuous tracking protocols, a tool that worked perfectly during a pilot in January might give flawed recommendations during an inspection in November.Poor Data Lineage and Security
Where does the information go when you type it into a generic tool? Does the vendor use your proprietary molecule data to train their public models? Many basic applications lack clear data lineage. They cannot prove who had access to the data, how it was modified, or where it is stored. This violates fundamental data validity principles that require all records to be fully traceable and secure.
The Core Pillars of True AI Governance
Transitioning from an experimental sandbox to a validated environment requires a formal governance structure. Organizations must stop treating advanced tools as simple IT upgrades and start treating them as highly regulated assets. True governance rests on five core pillars.

Pillar 1: Use Case Intake and Risk Classification
You should not give every department open access to activate advanced tools whenever they want. A mature company implements a formal intake process. Before a single line of code is written or a vendor software is purchased, the business must capture the exact purpose, ownership, and expected benefit of the tool.
The tool must be classified by risk once captured. A helpful framework divides applications into three buckets:
High Risk: Systems that support clinical decision making, patient safety, quality control inspections, or deviation management. These require absolute validation rigor, design controls, and intense testing.
Medium Risk: Tools used for operational forecasting, supply chain streamlining, or trend analysis. These require clear procedural controls and standard validation.
Low Risk: Systems used for simple productivity, basic grammar corrections, or internal meeting scheduling. These require basic security reviews but minimal validation.
By tying your compliance controls directly to the risk level, you avoid over-documenting low-risk tools while assuring high-risk applications are bulletproof.
Pillar 2: Data Controls and ALCOA+ Principles
Every piece of information used by an advanced system must comply with strict data-integrity guidelines. This means all data must be attributable, legible, contemporaneous, original, and accurate. It must also be complete, consistent, enduring, and available.
Purpose-built solutions enforce these principles by creating strong data boundaries. They restrict the software so it can only access approved, verified source systems. They block the tool from pulling random information from the public internet. Furthermore, the system must keep an immutable audit trail. Every single prompt, every generated response, and every user validation must be permanently stamped with a time, date, and user identity.
Pillar 3: Mandatory Human Review
No advanced system should operate completely on autopilot when product quality or human lives are on the line. Governance frameworks ought to mandate a qualified human reviewer to check the work.
The software acts as an assistant, not the final judge. If the tool drafts a response to a quality deviation, a trained quality professional must review the source data, verify the accuracy of the draft, and officially sign off on the record. The system must store this human verification as part of the permanent compliance history.
Pillar 4: Continuous Performance Monitoring
Because advanced software can shift over time, you need a preemptive strategy to catch errors before they reach an auditor. This involves formulating clear metrics for model exactness, sensitivity, and fault rates.
Organizations must run regular challenge tests. These tests feed the system known data sets to verify that it still produces the expected results. If the performance drops below some threshold, it must trigger an automatic alert. The tool is then taken offline or restricted until a change control process evaluates the issue and revalidates the configuration.
Pillar 5: Thorough Vendor Qualification
Most companies do not build advanced language models from scratch. They partner with IT providers or integrate specialized software into their operations. However, regulators hold you responsible for your vendors' compliance.
You must thoroughly audit your technology partners. You need to inspect their security measures, bias detection protocols, and change control processes. If a vendor pushes an unannounced software update that alters how the model reasons, your validated status could vanish instantly. You must use vendors that offer complete honesty and give you control over when updates are applied.
Applying Computer Software Assurance to Advanced Systems
The thought of validating a dynamic, learning model can terrify traditional quality assurance teams. If you try to apply old, paperwork-heavy computer systems validation methods to advanced software, you will quickly find yourself buried in endless documentation. A typical project could take eight months to complete, destroying your competitive advantage.
Fortunately, the regulatory domain has evolved. The finalized computer software assurance guidance provides a modern framework that aligns perfectly with advanced technology.
Computer software assurance flips the script on validation. Instead of spending eighty percent of your time writing exhaustive test scripts and twenty percent on critical thinking, this system tells you to spend most of your time on risk analysis and critical thinking. It allows teams to focus their testing energy on the specific functions that directly impact product quality and patient safety.
When you apply this approach to advanced technology, validation becomes manageable. Instead of testing every likely response the tool could ever generate, you focus on the workflow's configuration. You test the boundaries, the data connectors, the human review steps, and the failure modes.
Organizations that utilize this risk-based framework see massive improvements. Validation timelines can drop from several months to just a matter of weeks. This allows life sciences companies to deploy powerful, automated solutions quickly without sacrificing a single shred of regulatory compliance.
How Purpose Built Compliance Platforms Solve the Problem
Living in the gap between a pilot and a validated system is dangerous and expensive. It wastes time, frustrates engineers, and leaves your business exposed to severe regulatory penalties. The solution is to step away from generic tools and adopt systems built from the ground up for regulated environments.
This is where specialized platforms make a massive difference. For instance, companies planning to manage their complex operations turn to dedicated provider ecosystems like PSC Software. Instead of trying to force a consumer application to comply with strict global laws, organizations leverage platforms designed with compliance as a core feature.
When you look at the product offerings within the life sciences ecosystem, you can see how purpose-built tools bridge the gap. For example, managing the intense training demands of a regulated workforce requires absolute precision. Neither a manual spreadsheet nor a standard corporate training tool can withstand the pressure of an audit. Using an automated option like the ACE LMS software solution ensures that every training event, standard operating procedure update, and employee qualification is tracked inside an unalterable audit trail. This level of control perfectly aligns with the information-consistency standards required for advanced automation.
Traditional validation paperwork can slow an organization to a crawl. Converting to a digital, paperless environment using tools like ACE Validation's paperless GxP software allows teams to unify their compliance activities. With document control, corrective actions, and validation records live in a single, connected digital ecosystem, implementing and monitoring advanced technology becomes simple, enabling effortless tracking of data lineage, transparent management of system changes, and a clear, organized history for any inspector who walks through your door.
A Practical Roadmap to Inspection Readiness
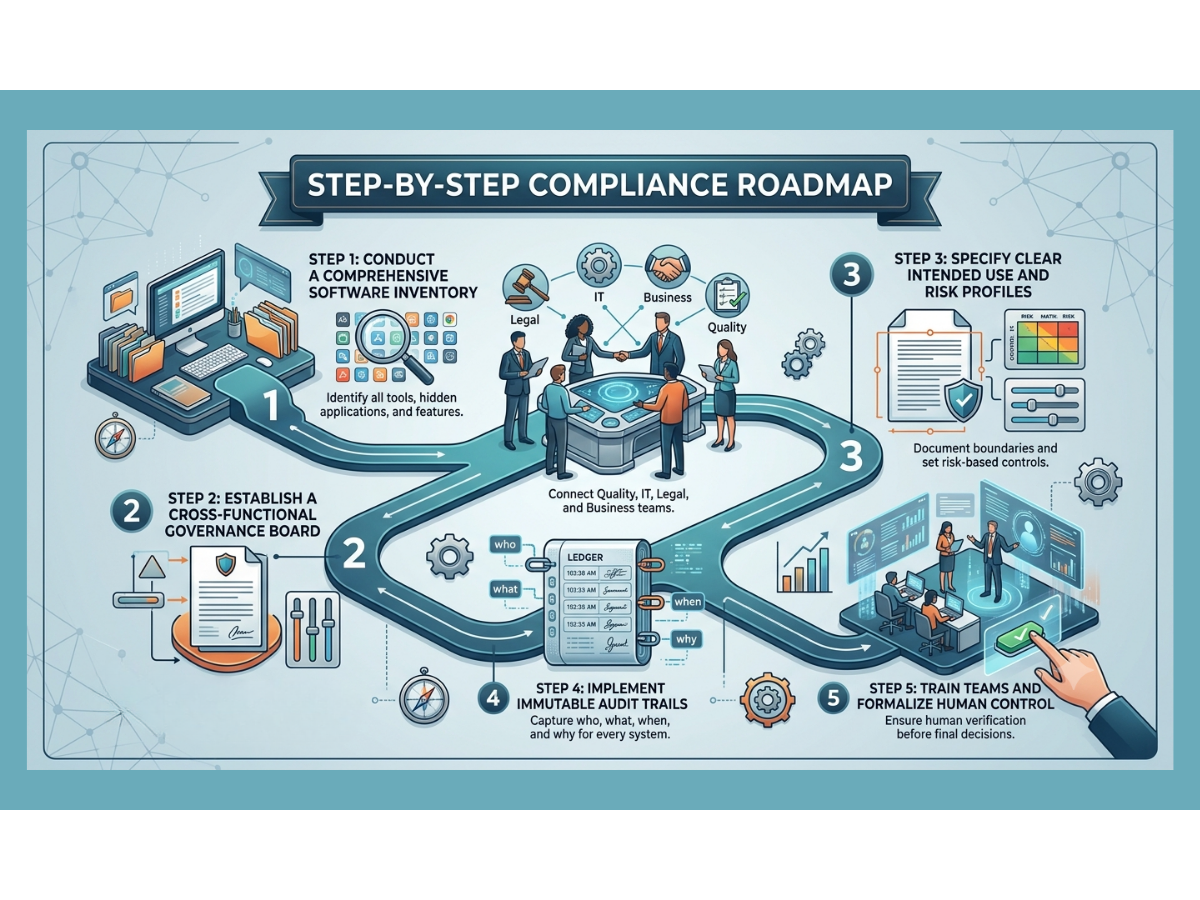
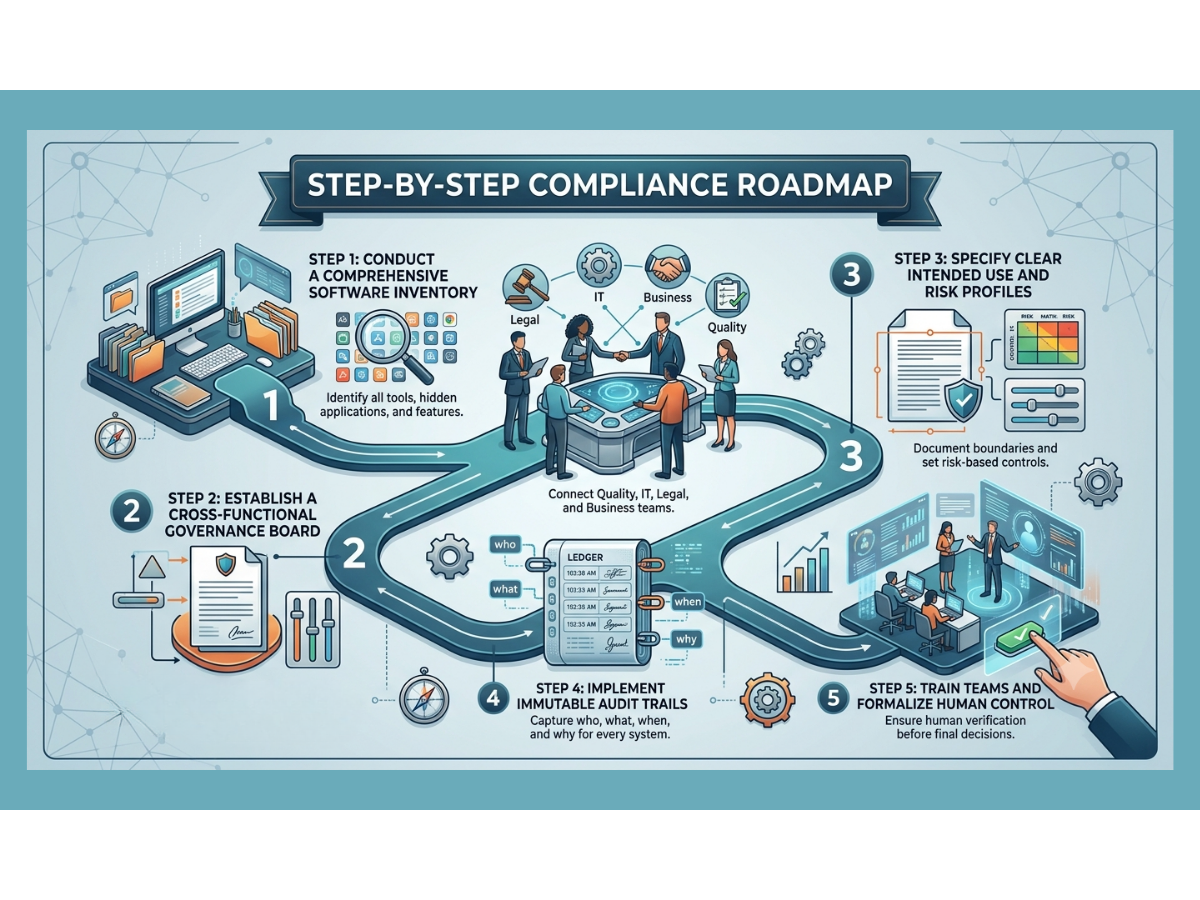
If your organization wants to close the gap and build artificial intelligence systems that are truly inspection-ready, you must follow a clear, step-by-step roadmap.

Step 1: Inventory All Advanced Tools
You cannot govern what you do not know exists. Conduct a thorough audit across your business to discover every tool currently in use. Look for hidden applications where employees might be pasting company data into public websites. Document every vendor-supplied feature that claims to use smart automation.
Step 2: Create an Internal Governance Board
Bring together leaders from quality assurance, information technology, legal, and operational business units. This group will serve as the gatekeepers for all automation projects. They will review new use cases, assign risk classifications, and confirm that no project moves forward without a clear validation plan.
Step 3: Draft Clear Intended Use Statements
For every approved tool, write a detailed statement explaining exactly what the software is allowed to do and what it is strictly prohibited from doing. Document the data sources, the human review workflows, and the exact records the system will generate.
Step 4: Enforce Technical Data Controls
Work with your IT team or software vendors to confirm that every system has strong access controls, data encryption, and unalterable audit trails. Verify that the system layout prevents automatic model updates without requiring formal change control.
Step 5: Establish Continuous Monitoring Standards
Create a schedule for regular performance reviews. Define your drift thresholds and write clear standard operating procedures for what the team must do if the software shows signs of declining accuracy.
Final Thoughts
Artificial intelligence offers incredible potential for the pharmaceutical industry and can help us analyze massive data sets, spot manufacturing deviations early, and streamline heavy documentation workloads. But these benefits mean absolutely nothing if the technology cannot survive a regulatory inspection.
The era of playing around with casual pilots is over. Regulatory bodies are stepping up enforcement, and the companies that succeed will be those that treat automation with the discipline it deserves. By shifting away from generic tools and deploying purpose-built software, sound risk frameworks, and complete data lineage, you can confidently move your technology out of the sandbox and into a fully validated, inspection-ready reality.
Bridge that gap between AI innovation and regulatory reality. Contact Metis Consulting Services today. We are experts who can streamline your computer software assurance, fortify your governance structure, and ensure your technology is fully validated and completely inspection-ready.

GLP and GCP: How the FDA and EPA Watch Over Science
Two of the most important sets of rules are called GLP and GCP. In 2026, the two main government agencies responsible for these rules, the Food and Drug Administration (FDA) and the Environmental Protection Agency (EPA), are working together more than ever before. Re: FDA & EPA Oversight.

This week in the Guardrail…
We explore how federal oversight is shifting standards in 2026, making rigorous data integrity the new baseline for every laboratory and clinic.
By Michael Bronfman
The world of making new medicines and chemicals is a very busy place. Every day, scientists are working in labs and clinics to find the next big cure or a safer way to clean our homes. Because these products can affect our health and the planet, the government has very strict rules to ensure everything is done correctly. Two of the most important sets of rules are called GLP and GCP. In 2026, the two main government agencies responsible for these rules, the Food and Drug Administration (FDA) and the Environmental Protection Agency (EPA), are working together more than ever before. This “shaking out” of the rules is changing how companies operate.
What Do These Letters Mean
To understand the future of science, you first have to know what these abbreviations stand for. They are like a specialized language for safety and honesty.
GLP stands for Good Laboratory Practice. These rules are for the early stages of research. This is the work that happens in a lab with test tubes, plants, or animals before a human ever touches the product. The Environmental Protection Agency uses these rules to ensure that a new pesticide or powerful cleaner will not harm the environment or the people using it.
GCP stands for Good Clinical Practice. These rules start when the research moves into a clinic and involves human volunteers. The Food and Drug Administration uses GCP to ensure that participants in medical studies are safe and that the results are truthful.
The FDA and the EPA Joining Forces
In the past, these two agencies mostly stayed in their own separate worlds. If a company were making a heart medicine, they would talk to the FDA. If they were making a new bug spray, they would talk to the EPA. But today, many new products fall into both categories. For example, a special soap that kills germs on your skin might be considered both a medicine and a chemical.
Because of this, the FDA and EPA are now sharing their notes. If an EPA inspector finds that a lab is messy or that the scientists are not recording their results correctly, they report it to the FDA. This means companies can no longer be “half good.” They have to follow the rules perfectly for both agencies. This coordination ensures that, no matter what kind of product is being made, the public is protected by the highest standards.Official Federal Register : How these agencies work together
Why Honesty Is the Only Policy
The main goal of both GLP and GCP is to ensure data integrity. Data is just a fancy word for the information and results gathered during an experiment. If a scientist says that a drug worked on ten people, the government wants to see the actual signatures and blood test results to prove it.
If a company is caught lying about its data, it can be fined millions of dollars. They might even be banned from ever making products again. This is why risk management is so important during the middle stages of a study. If a company finds a minor mistake in its lab records, it needs to fix it immediately. Waiting until later to “clean up” the paperwork is a huge risk that can lead to a total failure when the FDA or EPA comes to visit.
Protecting the People in the Studies
While GLP protects the science in the lab, GCP protects the people in the clinics. Every person who joins a clinical trial is a volunteer. They are doing something brave to help others. GCP rules ensure that these volunteers are treated with respect.
Under these rules, every volunteer must sign a form acknowledging the risks. This is called informed consent. The doctors must also closely monitor the volunteers for any side effects. If a patient experiences a headache or dizziness, it must be recorded in the official files. TheNational Institutes of Health provides extensive information on how these rules help keep people safe in medical research.
The Quality Control Revolution
In 2026, many companies are hiring specialized teams just to ensure quality. These teams are like the “referees” of science. They do not do the experiments themselves. Instead, they monitor the other scientists to ensure they follow all GLP and GCP rules.
They check that lab machines are properly calibrated. They check to ensure that every signature on a form is genuine and dated correctly. This might sound like a lot of extra work, but it saves the company from failing an inspection. When a company has a high “quality score,” the FDA and EPA can trust their results much more easily. Organizations like theSociety of Quality Assurance help train these specialized workers to stay up to date on the latest rules.
Using Technology to Stay Safe
Technology is making it easier for the FDA and EPA to do their jobs. In the old days, inspectors had to look through thousands of paper files. Now, most of the data is digital. This allows the government to look at the data in real time.
If a lab in California runs a test, an official in Washington, D.C., can see the results almost instantly. This helps catch mistakes before they become big problems. It also makes it harder for anyone to change their results later to make a drug look better than it really is. This transparency is a big part of why the “shake out” between the two agencies is happening so fast. Digital tools make it impossible to hide in the shadows.
The Global Impact of These Rules
Australia, Europe, and the United States all have their own versions of these rules. However, they are all starting to look very similar. This is good news for the public. It means that a drug tested in Australia can be sold in America as long as it follows the same high standards of GLP and GCP.
When countries agree on the rules, medicines can travel around the world much faster. This helps people in every country get the help they need without waiting years for additional testing. TheWorld Health Organization works to help all countries follow these same high standards for health and safety.
Education Is the Key
For these rules to work, every person in the pharmaceutical industry needs to be educated. It is not just the boss's job. Even the person cleaning the lab or the nurse at the clinic needs to understand why the rules matter.
Education helps people understand that following the rules is about more than just avoiding a fine. It is about making sure that the medicine your grandmother takes, or the soap you use on your children, is 100 percent safe. When everyone knows the “why” behind the rules, they are much more likely to follow them correctly every single day.
Risk Management and Quality Systems
Modern pharmaceutical companies use a Quality Management System (QMS) to track everything. A QMS is like a giant digital brain that stores all the company's rules and records. It helps with risk handling by flagging errors the moment they happen.
In 2026, risk management strategies are no longer just about fixing problems. They are about predicting them. By using clinical data management tools, a biotech firm can detect whether a machine is starting to wear out or whether a lab is experiencing too many small errors. This type of risk management planning helps prevent major disasters that lead to FDA warning letters.
The Role of REMS in Safety
Another way the FDA keeps people safe is through REMS, which stands for Risk Evaluation and Mitigation Strategies. These are extra safety programs for drugs that might be dangerous if not used exactly right. For example, some medicines require a patient to get a blood test every month. TheFDA REMS website tracks these programs to ensure drug companies are doing their part to manage risk.
Final Thoughts on the Future of Quality
The “shaking out” of rules between the FDA and the EPA is a positive step for everyone. It means that science is becoming more open and more honest. Companies are learning that they cannot take shortcuts during any stage of research.
By following the rules of GLP and GCP, we ensure that the future of medicine is bright. We can trust the products we buy because we know the government is watching and the scientists are being careful. Quality is not just a set of letters; it is a promise to the public that their safety is the most important thing of all. Don’t wait for a problem to appear before you start being careful. Start with quality on day one and the rest of the journey will be much smoother for everyone.
Frequently Asked Questions: GLP, GCP, and Regulatory Compliance
What is the main difference between GLP and GCP?The biggest difference is when they are used. GLP is for the preclinical stage, which is work done in a lab on animals or cells. GCP is for the clinical stage involving human volunteers.
Why does the EPA care about laboratory rules?The EPA ensures that chemicals like weed killers do not pollute our water. They use GLP to ensure companies are honest about chemical safety. You can find guidelines on theEPA Compliance Monitoring page.
Can a company fail a trial if they follow the science but miss the paperwork?Yes. In the eyes of the FDA, if a test was not documented correctly, it never happened. “Clean data” is just as important as the medicine itself.
What happens during an FDA or EPA inspection?Inspectors check original notebooks, computer logs, and even equipment logs. They want to see a clear trail from the study's start to the final report.
How does Risk Mitigation help with these rules?It means finding small mistakes before the government does. Fixing a mistake early in a study costs much less than fixing it later, when thousands of people are involved.
Where can I stay updated on these changing rules in 2026?The best place to watch for updates is theFDA Voice blog. This site tracks how the agencies are joining their rules for digital data. https://www.fda.gov/news-events/fda-newsroom/fda-voices.
The Pre-Inspection Compliance Checklist
When an inspector arrives, they look for a “culture of quality.” If you are preparing for an audit in 2026, here are the top ten things you must have ready.
Item
Description
1. The Master Schedule
A list of every study that has happened in your lab.
2. Current SOPs
Proof that your team is using the newest versions of your rulebooks.
3. Training Logs
Proof that every person was taught how to do their job before starting.
4. Equipment Records
Logs showing that every scale and fridge is checked regularly.
5. Chain of Custody
A record of who touched a sample at every single minute.
6. Raw Data
Original handwritten notes or machine printouts.
7. QA Reports
Reports from your own internal “referees.”
8. CAPA Plans
Records showing how you found and fixed past mistakes.
9. Computer Validation
Proof that your digital data is secure and cannot be changed.
10. Signatures and Dates
Every page must be signed with no blank spaces or whiteout used.
Final Tip: The Clean Room Rule
An inspector will also look at your physical space. If a lab is cluttered, they will assume the data is also messy. A clean and organized lab tells the inspector that you take quality management seriously. For more help, you can check the FDA Inspection Guide for the latest standards.
Secure Your Future in Science with Metis Consulting Services
When "good enough" no longer passes the test, your organization needs to turn regulatory pressure into a competitive advantage. Contact Metis Consulting Services today to ensure your next breakthrough is backed by an unbreakable foundation of compliance.

Understanding the Significance of CRLs Being Released: Beyond the Regulatory Language
The FDA's Complete Response Letter (CRL)-- few documents hold as much weight in the complex and often opaque world of pharmaceutical development, as the CRL. Metis Consulting can help navigate them, learn more at our blog, The Guard Rail.
Written by Michael Bronfman, July 28, 2025

This week in The Guard Rail, we at Metis are looking at a hot topic for our industry. Michael Bronfman tackles a hidden power in the pharmaceutical and medical device manufacturing world: the FDA's Complete Response Letters (CRLs). These are not just dry documents. The contents have traditionally been kept secret, known only to the receiving company. However, that secrecy might now be coming into the open. Why? Because a CRL can instantly derail a company's future, send stock prices plummeting, and, most critically, determine if a life-saving treatment ever sees the light of day. Join us as we uncover why these once-confidential letters are at the heart of a tidal wave push for transparency.
The FDA's Complete Response Letter (CRL)-- few documents hold as much weight in the complex and often opaque world of pharmaceutical development, as the CRL. For many outside the industry, the term might sound dry, bureaucratic, or even cryptic. But for drug developers, investors, patients, and clinicians, CRLs are pivotal turning points; letters that can reshape company strategy, impact stock prices overnight, and, most importantly, influence when or even if a new therapy reaches patients.
Historically, the contents of CRLs have often remained confidential, known only to the company receiving them and occasionally, selectively disclosed to the public. Yet the idea of CRLs being more broadly released, whether voluntarily by sponsors or systematically through policy change has gained traction. Why? Let us explore why these letters matter, what they contain, and why making them public can be a significant step forward for science, business, and patient trust.
What exactly is a CRL?
A Complete Response Letter is issued by the U.S. Food and Drug Administration (FDA) when it completes its review of a New Drug Application (NDA) or Biologics License Application (BLA) but decides not to approve it in its current form. Importantly, a CRL does not mean the drug is permanently rejected. Instead, it outlines the deficiencies that prevent approval and often provides guidance on what the sponsor could do to address them.
Deficiencies can include:
Issues with clinical efficacy or safety data (e.g., not enough evidence that the drug works, or safety concerns in certain patient populations)
Manufacturing or quality control shortcomings
Problems with labeling or risk management strategies
Statistical or methodological issues in trial design
For sponsors, receiving a CRL is both a setback and a roadmap. It’s an official document telling them: “Here is what is missing; come back when you have fixed it.”
The FDA's Complete Response Letter (CRL) few documents hold as much weight, in the complex and often opaque world of pharmaceutical development, as the CRL. For many outside the industry, the term might sound dry, bureaucratic, or even cryptic. But for drug developers, investors, patients, and clinicians, CRLs are pivotal turning points; letters that can reshape company strategy, impact stock prices overnight, and, most importantly, influence when or even if a new therapy reaches patients.
Historically, the contents of CRLs have often remained confidential, known only to the company receiving them and occasionally, selectively disclosed to the public. Yet the idea of CRLs being more broadly released — whether voluntarily by sponsors or systematically through policy change — has gained traction. Why? Let's explore why these letters matter, what they contain, and why making them public can be a significant step forward for science, business, and patient trust.
What exactly is a CRL?
A Complete Response Letter is issued by the U.S. Food and Drug Administration (FDA) when it completes its review of a New Drug Application (NDA) or Biologics License Application (BLA) but decides not to approve it in its current form. Note, a CRL does not mean the drug is permanently rejected. Instead, it outlines the deficiencies that prevent approval. Often, the letter will provide guidance on what the sponsor can do to address the issue.
Deficiencies can include:
Issues with clinical efficacy or safety data (e.g., not enough evidence that the drug works, or safety concerns in certain patient populations)
Manufacturing or quality control shortcomings
Problems with labeling or risk management strategies
Statistical or methodological issues in trial design
For sponsors, receiving a CRL can be a setback, but it is also can be a roadmap. It is an official document that says: "Here is what is missing; come back when you have fixed it."
Why are CRLs so important?
CRLs carry enormous significance because they sit at the intersection of science, business, and public health. Consider:
1. Strategic pivot points for companies
A CRL forces a company to decide: Do we invest more time and money to address the FDA's concerns, or do we walk away? Sometimes the deficiencies are minor and easily fixable; at other times, they are so fundamental that continuing to do so makes little sense.
2. Market-moving disclosures
Because the market places great value on new product approvals, the news of a CRL often leads to sharp drops in a company's stock price — especially if the drug was seen as a major pipeline asset.
3. Impact on patients
For patients waiting for new treatment options, CRLs can feel like an unexpected delay. Understanding the nature of the deficiency can help patients and advocates see whether it is a temporary hurdle or a sign of deeper problems.
4. Scientific learning
Each CRL is a detailed FDA critique of a drug's data and the sponsor's responses. While usually kept confidential, if shared, they can become case studies that improve drug development as a whole.
The current situation: Confidential by default
Under U.S. law, CRLs are part of a company's regulatory correspondence and thus are treated as confidential commercial information. Sponsors may choose to disclose the fact that they received a CRL — and often do, given that it's material information for investors — but the actual content is rarely released in full.
Instead, companies often issue press releases summarizing the FDA's concerns. Unfortunately, these summaries can be selective, vague, and overly optimistic:
Selective: emphasizing easily fixable manufacturing issues and omitting more serious efficacy concerns
Vague: using language like "additional analyses requested" without context
Optimistic: framing the CRL as "a minor setback" even if the letter itself is more critical
This practice makes it hard for outside observers — including investors, clinicians, and patient groups — to understand what really happened.
The significance of CRLs being more publicly released
CRLs regularly released in full, could have a profound effect on how new therapies are evaluated, understood, and debated. Here's why:
1. Transparency builds trust
Our industry struggles with perceptions of secrecy. Polished summaries are shared and that is fine but if they are the only data released, it is impossible to know if the sponsor is downplaying serious concerns. Releasing more complete CRLs shows the unfiltered FDA perspective, which can reassure the public that approvals are based on thorough, science-driven review.
2. Better information for stakeholders
Investors could better assess the real risk of resubmission and approval. Clinicians could understand why certain drugs were not approved — whether due to safety concerns in specific populations or inadequate evidence of benefit. Patients and advocacy groups could advocate more effectively if they knew the precise barriers.
3. Industry-wide learning
Drug development is full of repeated mistakes: inadequate trial design, poor endpoint selection, underpowered studies, or manufacturing gaps. Public CRLs can serve as detailed case studies, allowing future sponsors to avoid similar pitfalls.
4. Accountability
Public CRLs help ensure that sponsors fully address the FDA's concerns before resubmitting, rather than trying to sidestep them with minimal new data. They also keep the FDA accountable, making its reasoning transparent and open to scientific debate.
Potential drawbacks and industry concerns
Of course, releasing CRLs is not without controversy. Key concerns include:
1. Proprietary data
CRLs often contain detailed discussion of clinical trial data, manufacturing processes, and commercial plans. Sponsors argue that full disclosure could benefit competitors or harm competitive advantage.
2. Misinterpretation
FDA reviews are technical documents, and taken out of context, statements in a CRL could be misread by the public or sensationalized by the media.
3. Chilling effect on communication
If sponsors know that every word in their submissions could become public, they might be less candid, potentially limiting open dialogue with regulators.
4. Impact on innovation
Some fear that too much transparency could discourage small biotech firms — already operating under tight timelines and budgets — from pursuing high-risk programs.
The evolving conversation
The debate is not purely academic. In recent years:
Some sponsors have voluntarily released CRLs, especially when the market reaction to vague summaries was worse than anticipated.
Regulatory advocates and transparency groups have pushed for routine publication, arguing that CRLs, like European Public Assessment Reports (EPARs), could help demystify the approval process.
The FDA itself has signaled interest in improving transparency, though it is constrained by existing confidentiality laws.
The conversation reflects a broader trend in medicine: moving from "trust us" to "show us." Patients, payers, and clinicians want to see the data and the reasoning behind it, not just the headline.
International context
The U.S. FDA is not alone in grappling with this issue. European regulators, through the EMA, publish relatively detailed assessment reports once a drug is approved, but not if it is rejected. Similarly, Health Canada has taken steps to publish "Summary Basis of Rejection" documents for drugs that are not approved.
These models demonstrate that it is possible to balance transparency with the protection of confidential information, although it requires careful policy design.
A path forward
So, what would be the ideal outcome?
Routine publication of redacted CRLs: Share the FDA's reasoning while redacting truly proprietary data, like detailed manufacturing process steps.
Standardized summaries: Even if full letters aren't released, require sponsors to issue standardized, FDA-reviewed summaries that accurately reflect the deficiencies.
Educational context: Provide plain-language explanations alongside CRLs, so clinicians, patients, and journalists can understand the technical details.
Such steps could bring real benefits without undermining innovation.
Why it matters
At its heart, the significance of CRLs being released is about more than a document. It is about shining light on critical moments in the life of a new therapy: the point where data meets judgment. When companies keep those moments private, the public can only guess at what went wrong. When CRLs are shared, everyone from researchers designing the next trial to patients hoping for a breakthrough can see, learn, and plan accordingly.
Transparency is not a cure-all. It won't eliminate uncertainty, disappointment, or risk. However, in a field where trust is essential and decisions affect both lives and balance sheets, sharing the FDA's reasoning is a powerful way to build confidence, foster learning, and ultimately bring better medicines to the people who need them.
If your organization is grappling with CRLs or needs help avoiding them, please contact us at Metis Consulting Services: Hello@MetisConsultingServices.com.
For more info, see our website www.MetisConsultingServices.com

Matlock’s Misguided Take on the Opioid Crisis
The opioid crisis is a multifaceted public health crisis that has devastated communities around the world. This crisis is a result of a confluence of factors, including addiction, overprescribing, inadequate pain management education, and the illicit manufacturing and distribution of synthetic opioids. While opioids are a valuable tool for managing pain when used appropriately and under medical supervision, their misuse and abuse have led to widespread addiction, societal harm, and death by overdose.
Written by Dr. Olivia Fletcher

Kathy Bates' revival of the iconic Matlock series has brought back the legal drama we all know and love. As much as we love the reboot, the portrayal of the opioid crisis is deeply flawed and misleading. In the pilot episode, Madeline “Maddy” Matlock suggests the attorneys she works with had confidential client information that would have motivated the FDA to take opioids off the market ten years earlier. This claim is both inaccurate and harmful, as it misrepresents the complexities of the opioid crisis and disregards the ethical obligations of attorneys.
The Opioid Crisis: A Complex Issue
The opioid crisis is a multifaceted public health crisis that has devastated communities around the world. This crisis is a result of a confluence of factors, including addiction, overprescribing, inadequate pain management education, and the illicit manufacturing and distribution of synthetic opioids. While opioids are a valuable tool for managing pain when used appropriately and under medical supervision, their misuse and abuse have led to widespread addiction, societal harm, and death by overdose.
Attorney-Client Privilege: A Cornerstone of the Legal System
The principle of attorney-client privilege is fundamental in our legal system. It protects confidential communications, ensuring that individuals can seek legal advice without fear of disclosure. This privilege is essential for the establishment and maintenance of trust in the legal system to facilitate open and honest communication between attorneys and their clients. Attorney-client privilege only allows the attorneys to divulge communications in the case the client intends to use advice from the attorney to commit further crimes.
As a therapist myself, confidentiality and morality don’t always play nicely together. Confidentiality is the province of ethics, not morality. What may seem Right or Wrong in the micro may not align neatly with what is necessary in the macro. Our legal system requires clients to be able to confide fully in their attorneys in order to assist in their own representation. So while a client cannot ask an attorney for advice on how to hide the body of the next person they murder, they absolutely can tell the attorney they plan to murder someone else and the attorney’s hands are tied. This is the strictest confidentiality in our legal system. No other communication is given such broad protections by our courts. Therapists, physicians, clergy, and even spouses have more exceptions.
The Role of the FDA in Opioid Regulation
The Food and Drug Administration (FDA) plays a crucial role in regulating the safety and efficacy of prescription drugs, including opioids. The FDA approves drugs for use based on rigorous scientific evidence and requires manufacturers to submit post-market surveillance data to monitor the safety of approved products. If the FDA becomes aware of serious safety concerns, it may take action to restrict or withdraw the approval of a drug. As explained in the book, Empire of Pain by Patrick Radden Keefe, the Food and Drug Agents involved in monitoring Purdue Pharma, the company responsible for initially bringing oxycontin to the market, were deeply entwined with the company.
The REMS Program: A Critical Tool for Opioid Risk Management
The Risk Evaluation and Mitigation Strategy (REMS) program is a regulatory tool used by the FDA to manage risks associated with certain drugs. REMS programs can include requirements for prescribers, pharmacies, and patients, such as education and training, monitoring, and restrictions on distribution. Opioids are subject to REMS programs that aim to reduce the risk of abuse and diversion.
The Opioid Analgesic Risk Evaluation and Mitigation Strategy (REMS) is a program implemented by the FDA to address the risks associated with opioid analgesics. It requires certain actions from healthcare providers and patients to ensure the safe use of these medications.
The requirements of these programs include:
For Prescribers:
Education: All healthcare providers involved in the management of patients with pain, including prescribers, nurses, and pharmacists, must complete accredited continuing education (CE) courses on the safe use of opioids. These courses must be based on the FDA's Opioid Analgesic REMS Education Blueprint.
Patient assessment: Healthcare providers must assess a patient's risk factors for opioid misuse, addiction, and overdose before prescribing opioids. This assessment should include a thorough medical history, physical examination, and consideration of the patient's social and psychological factors.
Treatment plan: If opioids are prescribed, healthcare providers must develop a comprehensive treatment plan that includes:
Clear goals for pain management
A risk assessment and mitigation strategy
A plan for monitoring the patient's response to treatment
A plan for tapering or discontinuing opioids as appropriate
Documentation: Healthcare providers must document their assessment, treatment plan, and monitoring of the patient's response to treatment in the patient's medical record.
For patients:
Education: Patients who are prescribed opioids must receive information about the risks and benefits of these medications, as well as instructions on how to use them safely.
Agreement: Patients must sign a written agreement that outlines their understanding of the risks and benefits of opioids and their commitment to follow the prescribed treatment plan.
Monitoring: Patients must be monitored regularly for signs of opioid misuse, addiction, and overdose. This may include regular follow-up appointments, urine drug testing, and other assessments as appropriate.
Manufacturer requirements:
Manufacturer reporting: Opioid manufacturers must report any adverse events associated with their products to the FDA.
Public education: Opioid manufacturers must also conduct public education campaigns to raise awareness about the risks of opioid abuse and addiction.
Risk vs Benefit
All medications come with risk and side effects. Even the most thorough clinical trials may not make all of these clear in the limited time and population available. One of the key points the FDA focuses on is whether the benefits of the medication outweigh the risks. Midrin, a migraine abortant, was widely prescribed for decades before studies indicated that the Schedule IV substance, dichloralphenazone, which was one of three substances in midrin, was too prone to abuse. That combined with the concerns over liver damage from overuse of acetaminophen led the FDA to remove it from the market. Opioids do provide significant pain relief to patients for whom they are prescribed. It is important not to throw the baby out with the bath water. As we continue to advance pharmaceutical science, we may, hopefully, find a way to relieve pain without the dangers associated with narcotics. Knowing everything we know about the dangers of opioids, they are still considered worth the risk.
Conclusion
While Matlock is an entertaining show, the portrayal of the opioid crisis is deeply flawed and highly misleading. The show's suggestion that attorneys could have prevented the crisis by divulging confidential client information is inaccurate and disregards the ethical obligations of attorneys. The opioid crisis is a complex issue that requires a multifaceted approach, including improved pain management education, increased access to treatment, and stricter regulations on the prescription and distribution of opioids. We have enough misinformation circulating without adding to the detritus, perhaps some accountability is due here.