

AI in Regulatory Submissions: Writing for Both Human and Machine Reviewers
This week in the Guardrail, we analyze the dual-audience reality facing modern pharmaceutical compliance. As regulatory agencies integrate automated tools to parse complex submissions, drug sponsors must adapt their documentation strategies to satisfy both algorithmic logic and human expertise.

This week in the Guardrail, we analyze the dual-audience reality facing modern pharmaceutical compliance. As regulatory agencies integrate automated tools to parse complex submissions, drug sponsors must adapt their documentation strategies to satisfy both algorithmic logic and human expertise.
By Michael Bronfman
May 25, 2026
The world of making and approving medicines is going through a massive shift. For decades, pharmaceutical companies wrote drug applications for just one audience: human scientists. Teams of medical doctors, chemists, and statisticians at agencies like the Food and Drug Administration would read thousands of pages of text to decide if a new drug was safe.
Today, that process looks very different. Pharmaceutical companies now use computer algorithms, known as Artificial Intelligence, to run clinical trials and analyze data. At the same time, the regulatory agencies themselves are starting to use computer programs to help read and sort through massive piles of application documents.
This means medical writers and drug sponsors must now write for two very different audiences at the same time. They must write for the human experts who make the final decisions, and they must write for the machine reviewers who scan the text for errors and patterns. If an application is not structured correctly for a machine to read, it could get flagged for inconsistencies before a human expert even looks at it.
To help companies navigate this change, the Food and Drug Administration released official draft guidance about using these advanced computer models in drug development. This document outlines exactly how the agency looks at data generated by computers and how companies should share that information. For more detailed context, you can read the official announcement on the FDA Press Release Page.
The Food and Drug Administration Risk Framework
The official policy from the government makes one thing very clear: not all computer applications are treated equally. The agency uses a risk-based framework to grade how much scrutiny a system needs. This framework is based on two main ideas: model influence and decision consequence.
Model influence means how much the computer output affects the final decision. If a computer makes a final choice on its own, its influence is strong. If a human expert checks the work and can override the computer, its influence is lower. Decision consequence means what could go wrong if the computer makes a mistake. If a computer error harms a patient, the consequences are high. If an error just slows down a factory machine for an hour, the consequence is low.
By looking at these two factors, the government separates computer tools into high-scrutiny systems and low-requirement systems.

High Scrutiny Systems
The highest level of official review is saved for computer systems that directly create evidence for a drug application. These are systems where a mistake could directly hurt a patient or ruin the results of a scientific study.
The government pays closest attention to these five specific areas:
Patient Stratification: Choosing which patients get to be in a clinical trial based on their genetic codes or medical histories.
Dose Optimization: Using mathematical models to calculate exactly how much medicine a patient should take to get better without getting sick from side effects.
Real World Data Analysis: Scanning millions of electronic health records from hospitals to see how a drug performs in everyday life outside of a controlled trial.
Safety Signal Detection: Watching patient data in real time to spot rare and dangerous side effects before they become a widespread public health crisis.
Endpoint Derivation: Using wearable sensors like smartwatches to measure how well a patient is moving or sleeping during a clinical trial.
If a company uses a computer for any of these tasks, it must prove the system is incredibly reliable. They must show how the model was trained, what data it used, and how it avoids bias.
Low-Requirement Systems
On the other side of the coin, some computer uses do not impact patient safety at all. If a company uses a computer tool to format a document, check page numbers, or organize internal administrative tasks, the government does not need to see piles of validation data. These internal operations face proportionally lower requirements because a mistake by the computer will not change the scientific conclusions of the drug trial.
Understanding the Double Audience
Because regulatory agencies are now using advanced software to help manage incoming applications, drug sponsors must realize they are writing for a double audience. The text must satisfy both the human brain and the computer algorithm.
To see how these two audiences read differently, look at this comparison:
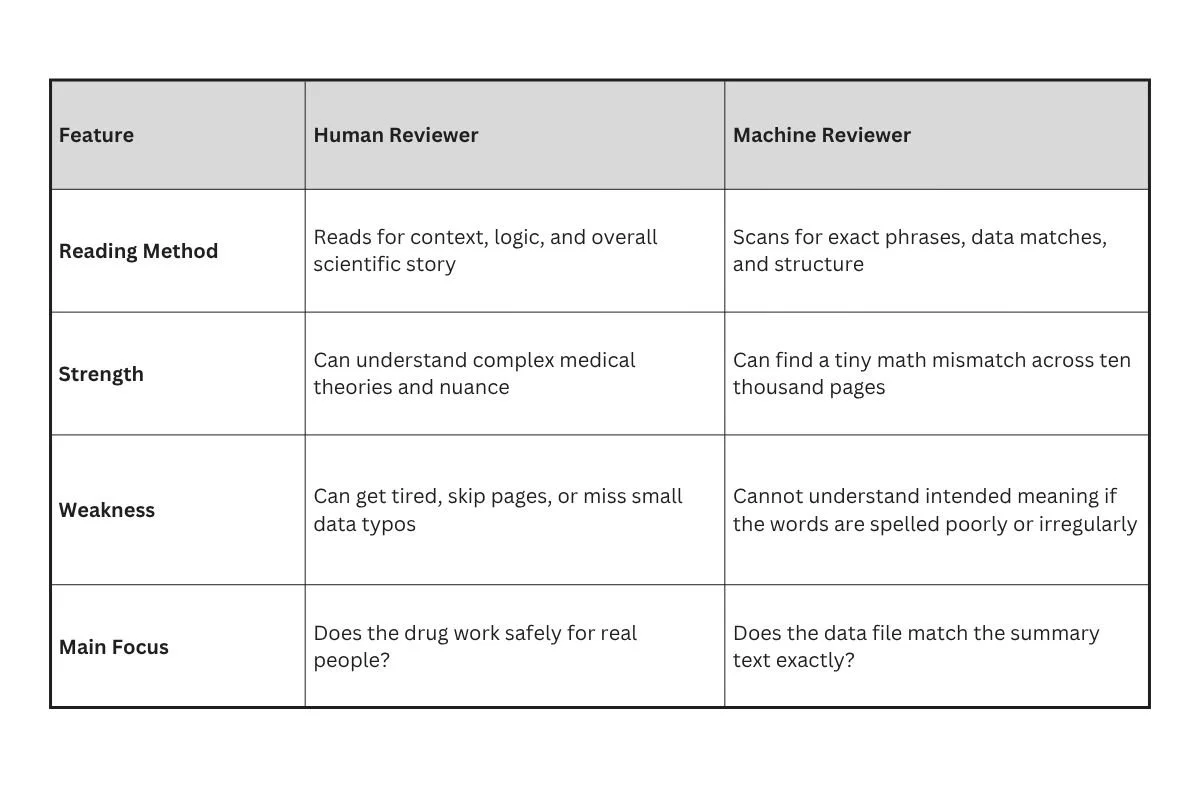
When a human reads a drug application, they want a clear narrative. They want to understand the journey of the drug from the laboratory to the clinic. They care about scientific logic.
A machine reviewer does not care about stories. It treats the document like a database. It looks at the tables, the labels, and the numbers to make sure everything adds up perfectly. If the summary on page five says fifty patients had a headache, but the raw data table on page nine hundred says forty-nine patients had a headache, the machine will flag that instantly. A human might miss that small slip, but a machine never will.
Writing for the Machine Reviewer
Writing for a computer means changing how you present text. Computers like clean organization, predictable patterns, and explicit language. If you write with vague words, the software can get confused and flag your document as a risk.
Structure and Predictability
The best way to help a machine reviewer is to use standard templates. Regulatory documents should follow strict structural rules. Use clear, standardized headings for every section. Do not try to be creative with section titles. If the standard title is Clinical Efficacy, do not change it to How Well the Drug Worked. The computer looks for specific keywords to map the document, and changing those keywords breaks the map.
Data Consistency and Labels
Every data point must look identical throughout the entire file. If you refer to a drug concentration as ten milligrams on one page, do not write it as 10mg on the next page. Choose one format and stick to it.
Also, make sure that every chart and table has clear, descriptive labels that use text instead of scanned images. Machine reviewers read text characters, not picture pixels. If you paste a picture of a table into your document, the computer sees a blank space and misses all the important data inside it.
Front Loading for Clarity
Machines are built to look for core conclusions early. Put your main findings, safety summaries, and essential data points right at the front of your sections. Do not hide your main message under paragraphs of introductory fluff. Front loading your clarity helps the computer categorize your document correctly on its very first pass.
Writing for the Human Reviewer
While you must make your document easy for a computer to analyze, you cannot forget the human being who must sign the final approval paper. Humans need context, clear explanations, and a believable scientific argument.
Explaining the Why
A machine can show that a number changed, but only a human can explain why it changed. If a clinical trial had a sudden drop in patient attendance during month four, a machine might flag it as a data error.
The human writer needs to explain the context:
"Patient attendance dropped in month four due to a historic blizzard that closed three major clinical trial sites for two weeks, but patients resumed their regular visits as soon as the roads cleared."
This explanation satisfies the human reviewer and prevents them from rejecting the data.
Keeping the Story Alive
A good regulatory submission tells a story of safety and success. The human writer must connect the dots between different pieces of research. Show how the animal studies predicted the human results, and show how the human results match the goals of the project. Use active, plain verbs to explain what the scientists did. Avoid overly dense language that puts the reader to sleep. A tired reviewer is a frustrated reviewer.
Conducting an Internal Review
Before you click the submit button to send your drug application to the government, your team should perform a complete internal review. This means testing your document against your own software tools to see what a machine reviewer will find.
Step One: The Automated Consistency Check
Run your completed document through text-matching software. This program should look for every number, percent, and statistical value to make sure they match perfectly across all chapters. If the software finds a conflict, fix it immediately. You want to find these errors yourself rather than letting the government find them first.
Step Two: The Structure Audit
Verify that every hyperlink works and leads to the correct appendix. Check that your document map functions properly and that all headings match the standard table of contents. If a machine cannot navigate your document links, it may automatically reject the file.
Step Three: The Human Readability Pass
Have a scientist who did not write the document read it for flow and clarity. Ask them if the arguments make sense and if the explanations are easy to find. This step ensures that once your document passes the computer gates, it will please the human experts.
The Path Forward for Drug Developers
The use of computer intelligence in regulatory submissions is not a temporary trend. It is the permanent future of medicine. Drug companies that learn how to write for both humans and machines will get their medicines approved much faster. Those who stick to old ways of writing will face constant delays, data flags, and rejection notices.
To keep up with these changes, companies should train their medical writers in basic data science principles. Writers do not need to learn how to code, but they do need to understand how computers read and sort information. By focusing on predictability, exact data matches, and clear summaries, you can create a document that satisfies the cold logic of a machine and the deep wisdom of a human scientist.
To learn more about how the government views these new digital tools, you can review the comprehensive resources provided by theFDA Artificial Intelligence Development Page. Staying informed about these official updates is the best way to ensure your future submissions are successful.

FDA Under DOGE: What Workforce Reductions Mean for the Pharmaceutical Industry
FDA Under DOGE: What Workforce Reductions Mean for the Pharmaceutical Industry

There are seismic shifts occurring within the FDA as DOGE-led workforce reductions redefine the boundaries of regulatory oversight. It is a new era where the burden of pharmaceutical safety is shifting from the government to the private sector.
By Michael Bronfman
May 18, 2026
American healthcare is undergoing a massive shift in 2026. Under the new Department of Government Efficiency (DOGE), the Food and Drug Administration (FDA) has faced a notable transformation. More than 3,500 employees have been let go. These aren't just office workers; they are the scientists, inspectors, and experts who make sure the medicine in your cabinet is safe.
For people working in the pharmaceutical industry, this is a "mission-critical" moment. When the government agency that watches over you loses a large chunk of its workforce, the rules of the game change. You have to understand what a "leaner FDA" means for your daily job and for the patients who count on your products.
The Scale of the Change
To understand the impact, we have to look at who is gone. The cuts have hit almost every part of the agency. We are seeing fewer people in charge of:
Approving drug labels: Making sure the instructions on a bottle are correct and easy to read.
Posting recall notices: Getting the word out quickly when a dangerous product is found.
Testing samples: Actually looking at the chemicals in a lab to verify they match the given recipe.
According to Healthgrades reports, these cuts are already being felt on the ground. When you lose that many people, the wait time for everything starts to grow.
The Ripple Effect on Inspections
In the past, pharmaceutical companies expected regular visits from FDA inspectors. These visits kept everybody on their toes. With a smaller workforce, the FDA cannot be everywhere at once. Currently proposed is a “one-day inspection,” which may not be sufficient time for a regulatory body to carry out a thorough inspection of patient-facing treatment.
Legal experts at Ropes & Gray LLP have noted that workforce reductions will likely lead to longer investigative timelines. If there is a problem at a facility, it might take much longer for the agency to find it, or to clear a company that has resolved an issue. This creates significant uncertainty for sponsor organizations.
The Impact on Global Trade
The FDA doesn't just watch over US manufacturing sites. They also inspect sites globally, including in India and China, that export medicine to the United States.
International Inspections
Travel is expensive and time-consuming. With fewer inspectors, the number of overseas visits has dropped sharply. This creates a risk. If an overseas plant knows it won't be inspected for 5 years, it might get lax about its standards.
Smart companies are now performing their own "Supply Chain Audits." They are sending their own teams to visit their partners worldwide to ensure that every ingredient is pure. You cannot afford to have a partner who cuts corners.
Navigating Internal Reorganizations
The FDA is also being reorganized. Offices are merging, and departments are being renamed. For a pharma company, this means your "point of contact" might change every month.
Tips for Staying Connected
Document Everything: Keep a clear trail of every email and phone call with the agency.
Be Clear and Concise: Since FDA staff are overwhelmed, make your letters easy to read. Use bullet points and put the most important info first.
Monitor the Federal Register: Stay updated on new rules being issued to address the smaller workforce.
The Economic Reality
DOGE’s goal was to save taxpayers' money. While the government is spending less on salaries, the pharma industry might end up spending more.
The industry is learning that "less government" doesn't always mean "less work." It often means the work stops in the approval pathway.
Looking Ahead: The Future of the FDA
The year 2026 will be remembered as a turning point. We are moving toward a model in which the government sets the high-level rules, while companies are expected to police themselves much more strictly.
A New Partnership
The relationship between the FDA and pharma companies used to be like a teacher and a student. The teacher (FDA) would grade the student’s (Pharma) work and tell them how to fix it.
Now, the relationship is more like a judge and a citizen. The judge doesn't have time to teach you. They only have time to show up when something goes wrong and hand out a punishment.
Practical Compliance Steps for 2026
If you want to survive and thrive in this new environment, your team should focus on these three pillars:
1. Data Integrity
Every number in your report must be perfect. Since the FDA will be doing fewer "random checks," they will likely be much harsher when they find a data error. They will assume that if you made one mistake, you are hiding others.
2. Supply Chain Transparency
Know exactly where your Active Pharmaceutical Ingredients (APIs) come from. If your supplier in another country hasn't seen an FDA inspector in three years, you need to be the one to inspect them.
3. Rapid Response Teams
Have a plan ready for when something goes wrong. If you find a safety signal, you need to know exactly how to handle a recall without waiting for the FDA to hold your hand through the process.
The New Mission
The workforce reductions at the FDA are a challenge, but they are also an opportunity. Companies that prove they can maintain high standards without constant government supervision will win the trust of doctors and patients.
For pharmaceutical professionals in quality and regulatory , the mission is the same, but the pressure has increased. You are now the primary protectors of public health. By staying informed through resources like Healthgrades and keeping an eye on legal shifts at sites like Ropes & Gray LLP, you can navigate this leaner landscape more confidently.
The FDA might have fewer people, but the patients still expect the same level of safety. It is up to us to deliver it.
Is your organization ready for a one-day inspection or a supply chain failure? Discover the gaps in your compliance strategy; contact Metis Consulting Services today to fortify your quality systems and navigate the leaner regulatory landscape of 2026 with confidence.
Key Takeaways
The FDA is smaller now. Over 3,500 people lost their jobs, meaning the government has fewer experts to monitor the Pharma field
Wait times are longer. With fewer workers, the FDA may take longer to approve new drugs or investigate safety issues.
Companies have to watch themselves. Since our "government watchdog" is busy, drug companies must hire their own experts to ensure their medicines are safe.
Quality is more important than ever. If a company makes a mistake, it might have to handle cleanup on its own, with little help from the government.
Safety is still the goal. Even with a smaller FDA, making sure patients are safe is the main goal.