

Regulatory Pathways FDA and EMA – Are You Prepared for Ongoing AI Supervision?
Regulatory pathways in the United States and Europe are becoming more complex. The FDA and the EMA continue to raise expectations for data quality, transparency, and oversight. At the same time, regulators are expanding their use of advanced digital tools, including artificial intelligence, to review submissions, monitor compliance, and identify risk.

As regulators deploy advanced digital tools to scan for inconsistencies in real-time, pharmaceutical companies must redefine their approach to data integrity and organizational transparency to stay ahead of the curve. This week, the Guardrail analyses how the FDA and EMA are transitioning from milestone-based reviews to the new model of continuous AI-driven oversight.
By Michael Bronfman, for Metis Consulting Services
February 16, 2026
Regulatory pathways in the United States and Europe are becoming more complex. The FDA and the EMA continue to raise expectations for data quality, transparency, and oversight. At the same time, regulators are expanding their use of advanced digital tools, including artificial intelligence, to review submissions, monitor compliance, and identify risk.
For pharmaceutical companies, this shift changes how regulatory readiness should be defined. It is no longer enough to meet written requirements alone. Companies must be prepared for continuous supervision supported by AI-driven systems that can detect patterns, inconsistencies, and signals faster than traditional reviews.
Understanding how FDA and EMA pathways work today and how AI supervision fits into them is essential for long-term success.
Core FDA and EMA Regulatory Pathways
The FDA and EMA share the same goal of protecting public health, but their regulatory pathways differ in structure and process.
In the United States, drugs are typically approved through the New Drug Application or Biologics License Application process. These submissions include clinical, nonclinical, and manufacturing data. The FDA evaluates whether the product is safe, effective, and manufactured under appropriate quality standards.
FDA drug approval information is available at https://www.fda.gov/drugs
In Europe, the EMA oversees centralized marketing authorization for many products. A single approval allows access to all European Union member states. The review is conducted by scientific committees that assess quality, safety, and efficacy.
EMA regulatory guidance can be found at https://www.ema.europa.eu
While the pathways differ, both agencies expect robust data, strong quality systems, and ongoing compliance after approval.
The Shift Toward Continuous Oversight
Historically, regulatory oversight followed clear milestones. Sponsors submitted data. Regulators reviewed it. Inspections occurred at defined points. Today, oversight is becoming more continuous.
Post approval commitments, real-world evidence, and ongoing safety reporting mean that regulators receive data throughout a product life cycle. AI systems allow agencies to process large volumes of information efficiently.
This means issues may be identified earlier and more frequently. Trends that once took years to surface can now be detected in near real-time.
How AI Is Used by Regulators
Regulators use artificial intelligence in several ways. These tools help prioritize reviews, flag anomalies, and focus inspections on higher risk areas.
For example, AI can analyze adverse event reports to identify safety signals. It can review clinical datasets for unusual patterns. It can also examine manufacturing data to detect deviations or data integrity concerns.
The FDA has published information on its digital transformation efforts.
The EMA is also investing in advanced analytics to support regulatory science and supervision. More information. While AI does not replace human judgment, it guides attention and speeds decision-making.
What This Means for Regulatory Submissions
AI supervision changes how submissions are evaluated. Inconsistent data, unexplained outliers, and poor documentation are easier to detect.
Sponsors must ensure that datasets are clean, traceable, and well explained. Narrative justifications should align with underlying data. Discrepancies between modules or sections can trigger questions.
Regulators may compare current submissions with historical data from the same sponsor. Patterns of issues across programs may influence review focus.
This makes consistency and standardization across submissions more important than ever.
Data Integrity Under AI Review
Data integrity has long been a regulatory focus. AI-driven oversight raises the bar further.
Systems that automatically scan data can detect missing values, duplicate entries, or unusual trends. Manual workarounds and undocumented processes are more likely to be noticed.
Sponsors should ensure that data governance is strong across clinical, manufacturing, and pharmacovigilance systems. Access controls, audit trails, and validation remain essential.
Preparing for AI supervision means assuming that data will be examined at scale and in detail. FDA data integrity guidance is available for reference.
Clinical Trial Data and AI Scrutiny
Clinical trial data is a major focus of regulatory review. AI tools can evaluate consistency across sites, subjects, and time points.
For example, unusually similar data across different sites may raise questions. Unexpected enrollment patterns or protocol deviations may be flagged.
Sponsors should strengthen monitoring and quality control during trials. Early detection of issues allows corrective action before submission.
Clear documentation of deviations and decisions is critical. AI may identify the issue, but human reviewers will expect clear explanations.
Manufacturing and Quality Oversight
Manufacturing data is another area where AI supervision plays a growing role. Process data, deviation reports, and change records can be analyzed to identify trends.
Repeated deviations, delayed investigations, or weak corrective actions may draw attention. AI can also compare performance across sites or products.
Companies should ensure that quality systems are proactive rather than reactive. Trending and root cause analysis should be meaningful and timely. The FDA quality system expectations are clearly outlined on their site. Strong quality culture supports both compliance and operational performance.
Pharmacovigilance and Safety Monitoring
Post-market safety surveillance generates large volumes of data. AI helps regulators process adverse event reports more efficiently.
Signals may be detected earlier, leading to faster regulatory action. Sponsors must ensure timely and accurate reporting.
Safety databases should be validated and monitored. Follow-up procedures must be consistent and documented. Preparedness means having clear roles, trained staff, and reliable systems.
Here is a good description of FDA pharmacovigilance requirements
Transparency and Traceability Expectations
AI supervision increases expectations for transparency. Regulators may ask how conclusions were reached and how data was managed.
Traceability from raw data to final conclusions is essential. This applies to clinical analyses, manufacturing decisions, and safety assessments.
Documentation should be clear and accessible. Teams should be able to explain decisions without relying on informal knowledge.
This level of readiness supports inspections and builds regulator confidence.
Organizational Readiness for Ongoing Supervision
Preparing for AI-supported oversight is not just a technical challenge. It is an organizational one.
Leadership must support investment in systems, training, and governance. Teams must understand that oversight is continuous, not episodic.
Cross-functional collaboration becomes more important. Issues in one area may affect regulatory perception across the organization.
Training programs should emphasize data quality, documentation, and accountability.
Engaging With Regulators Proactively
Open communication with regulators remains important. Early discussions can help clarify expectations and reduce risk.
Sponsors should be prepared to explain how data is generated, managed, and reviewed. Transparency builds trust.
Regulatory science is evolving. Staying informed about guidance updates and regulatory initiatives helps organizations adapt. 1 2
Looking Ahead
AI supervision is becoming a permanent part of the regulatory landscape. It allows regulators to oversee more products, more data, and more activities with greater efficiency.
For pharmaceutical companies, this means readiness must be continuous. Quality, consistency, and transparency are no longer just best practices. They are essential expectations.
Organizations that embrace this shift and strengthen their regulatory foundations will be better positioned to navigate FDA and EMA pathways with confidence
Don’t wait to discover the gaps in your data integrity or submission strategy. Metis Consulting Services provides the expert governance frameworks and guidance you need to ensure your organization is not just compliant, but competitive.
Contact: hello@metisconsultingservices.com to fortify your regulatory foundation and navigate the complexities of FDA and EMA pathways with total confidence.

Clinical Trial Optimization
Clinical trials are the backbone of drug development. They provide the evidence needed to show that a product is safe and effective. They also represent one of the largest investments a pharmaceutical company will make

This week in the Guardrail, we dig deeper into clinical trial optimization. This article breaks down the essential strategies—from patient-centric design to proactive compliance—required to navigate the high-stakes journey from protocol to regulatory approval.
By Michael Bronfman for Metis Consulting Services
February 9, 2026
Clinical trials are the backbone of drug development. They provide the evidence needed to show that a product is safe and effective. They also represent one of the largest investments a pharmaceutical company will make. As development costs rise and competition intensifies, optimizing clinical trials is no longer just a nice-to-have or a good idea; it is essential.
Clinical trial optimization means designing and running studies in a way that protects patients, meets regulatory criteria, controls cost, and delivers clear answers. It is about working smarter, not cutting corners.
Why Optimization Matters More Than Ever
Clinical trials take time. Phase 2 and 3 trials may last several years from the first patient enrolled to the final data analysis. Delays are common and expensive. Missed enrollment targets, protocol amendments, and site performance issues can add months or even years to a program.
Each delay raises cost and reduces the effective patent life of a product. In competitive markets, delays can also mean losing the first-mover advantage. Optimization helps reduce these threats by improving planning, execution, and oversight.
Regulators expect sponsors to design trials that are scientifically sound and ethical. Poorly designed trials waste time and expose patients to unnecessary risk. Optimized trials support both business targets and regulatory compliance.
Strong Protocol Design Is the Foundation
Every optimized trial begins with a strong protocol. The protocol defines the study objectives, endpoints, population, and procedures. Weak protocols are one of the most common causes of trial failure.
Common protocol issues include excessively complex procedures, unclear endpoints, and overly restrictive eligibility criteria. These problems slow enrollment and increase protocol deviations.
Sponsors who involve cross-functional teams early have a higher success rate. Ideally, clinical, pharmacovigilance, regulatory, biostatistics, operations, and quality would all review protocol drafts. Early feedback identifies risks before the trial begins.
The FDA provides guidance on clinical trial design and conduct.
Patient Centric Design Improves Performance
Patients are at the center of clinical research, yet many trials are designed with little consideration for patient burden. Long visit schedules, frequent procedures, and complex instructions can discourage participation.
Optimized trials consider the patient experience. Simplifying visit schedules, decreasing unnecessary procedures, and using explicit communication improve enrollment and retention.
Patient-focused drug development initiatives encourage sponsors to incorporate patient perspectives.
FDA resources on this topic are available here
When patients stay engaged, data quality improves, and timelines are more predictable.
Site Selection and Support Are Critical
Clinical sites play a major role in trial success. Selecting sites based only on past performance or relationships may lead to poor results. Sponsors who use objective criteria such as patient population, access, staffing levels, and infrastructure are more likely to succeed.
Once sites are selected, how is ongoing support managed? Clear training, attentive communication, and realistic expectations help sites perform well.
High-performing sites reduce protocol deviations and data queries. This lowers the monitoring burden and improves inspection readiness.
Enrollment Planning Requires Realism
Enrollment challenges are one of the leading causes of trial delays. Overly optimistic enrollment projections often fail to account for competing trials, complex eligibility criteria, and patient availability.
Optimized enrollment planning uses real-world data where possible. This includes understanding disease prevalence, standard of care, and referral patterns.
Sponsors should also plan for contingencies. Backup sites, flexible enrollment strategies, and regular performance reviews help keep trials on track.
Data Quality Must Not Be an Afterthought
High-quality data is necessary for regulatory approval. Data errors, missing data, and inconsistencies can delay submissions and trigger regulatory questions.
Optimization includes building data quality into trial processes. Clear case report forms, standardized procedures, and timely data review help prevent issues.
Risk-based monitoring approaches focus attention on the most critical data and processes. The FDA provides guidance on monitoring clinical investigations.
These approaches support efficiency while continuing compliance.
Compliance Coordination From the Start
Optimized trials are designed with regulatory expectations in mind. This includes alignment on endpoints, comparators, and statistical analysis plans.
Early interaction with regulators can help clarify expectations and reduce surprises later. Meetings such as pre-IND and end-of-Phase 2 discussions grant valuable feedback.
FDA meeting guidance is also available.
Coordination with European regulators is also important for global programs. EMA guidance on clinical trials is found here.
Designing trials that meet multiple agency expectations lowers the need for additional studies.
Managing Protocol Amendments
Protocol amendments are common but costly. Each amendment adds time, cost, and operational complexity. Frequent amendments may also raise questions during inspections.
Optimized programs focus on reducing avoidable amendments. This starts with a thorough protocol review and feasibility assessment before trial launch.
When amendments are necessary, clear documentation and training are critical. Regulators expect sponsors to understand why changes were made and how they were implemented.
Vendor Supervision and Responsibility
Most clinical trials rely on vendors such as contract research organizations, laboratories, and data management providers.
While vendors perform key tasks, sponsors remain accountable.
Optimization includes strong vendor selection and oversight.
Clear contracts, defined roles, and success indicators help manage expectations.
FDA gives guidance on sponsor responsibilities. Regular oversight meetings and issue tracking help resolve problems early. Inspectors often review vendor oversight during inspections, making this a vital area of focus.
Inspection Readiness Starts During the Trial
Clinical trial optimization supports inspection readiness. Regulators may inspect sites, sponsors, or vendors during or after a trial.
Optimized trials maintain complete and accurate documentation. Training records, monitoring reports, and issue resolution logs should be readily available.
A culture of quality helps teams respond confidently to inspections. Waiting until a submission is filed to prepare for inspection is too late.
Using Lessons Learned Across Programs
Each trial generates valuable lessons. Optimized organizations capture and apply these understandings across programs.
Post-trial reviews can identify what worked and what did not; these insights may advance future protocol design, site selection, and operational planning.
Continuous improvement helps organizations remain competitive in a challenging environment.
Gazing Forward
Clinical trial optimization is an ongoing effort. As expectations evolve and pressures increase, sponsors must persist in refining their plans.
Well-optimized trials protect patients, support regulatory success, and control cost. They also help organizations deliver therapies to patients faster and with greater confidence.
In a market where delays are costly and scrutiny is high, optimization is far more than a best practice. It is a necessity.
Don’t let trial complexities stall your breakthrough. In an industry where every day counts, Metis Consulting Services can help you get to a streamlined, successful clinical program. Contact Metis Consulting Services today to optimize your path to approval and bring life-changing therapies to market faster.

Why the US Leaving the World Health Organization is Short-Sighted
On 22 January 2026, the United States announced that it was formally withdrawing from the World Health Organization. Public health experts, analysts, scientists, and those of us who work in the field find this a dangerous decision that will jeopardize national and global security and is scientifically reckless.

This week in the Guardrail, what it looks like when a major player walks away from a seat at the global health table and what that power vacuum actually means for the pharmaceutical industry's bottom line. Especially for businesses trying to stay competitive in a connected world.
By Michelleanne Bradley and Michael Bronfman, Metis Consulting Services
February, 2, 2026
On 22 January 2026, the United States announced that it was formally withdrawing from the World Health Organization. Public health experts, analysts, scientists, and those of us who work in the field find this a dangerous decision that will jeopardize national and global security and is scientifically reckless.
Leaving the World Health Organization is more than a political decision. The consequences are practical, measurable, and deeply connected to health, safety, and a stable economy. This decision weakens disease surveillance, slows drug development, raises health risks, and reduces US influence at a time when worldwide cooperation is imperative.
What the World Health Organization Does
The World Health Organization coordinates global public health efforts. It tracks infectious diseases, sets international health standards, supports vaccination programs, and helps countries respond to emergencies, including pandemics, natural disasters, and outbreaks of emerging diseases.
The WHO's role in medicine, quality, and safety is significant. It runs global systems that monitor drug shortages, counterfeit medication, and adverse drug reactions. These systems support regulators like the US Food and Drug Administration and help pharmaceutical companies operate safely across borders.
The mission of the WHO is to promote health, keep the world safe, and serve the vulnerable.
The US needs the WHO as much as the WHO needs the US.
How WHO Membership Protects the US
Many in the US assume that global health work benefits only other countries. In reality, WHO programs are a first line of defense for the United States.
Disease outbreaks do not respect borders. Viruses travel by plane faster than governments can react. The WHO operates a global disease surveillance network that alerts countries to new threats early, giving US health agencies and pharmaceutical companies time to prepare diagnostics and treatments. Without direct access and influence, the United States risks slower warnings and less reliable information.
During outbreaks such as Ebola, Zika, and COVID-19, WHO data-informed US public health decisions and supported early research efforts.
Currently, US companies are world leaders in medical research and diagnostics, and the WHO is a massive buyer of those goods. In 2023 alone, the WHO purchased over $600 million in US products. When the US is an active member of WHO, contributing to the stability of the global health market, we help prevent mass economic shutdowns. Interruptions to the supply chain, like those that cost the US trillions of dollars during the COVID-19 pandemic, are a concern. Withdrawal from the WHO creates a leadership vacuum that our rivals will fill. With the US involved, we can be sure that global health standards, norms, and research agendas will be consistent with our national interests.
Historically, the US has provided expertise in eliminating diseases worldwide, including smallpox, and has brought polio to the brink of eradication. U.S.-funded programs targeting HIV/AIDS (PEPFAR), tuberculosis, and malaria rely on the WHO’s coordination to be effective. By disengaging from these efforts, we risk collapsing the infrastructure we have built. This can lead to a resurgence of diseases we have spent decades and billions of dollars fighting to suppress.
Impact on Pharmaceutical Research and Drug Development
The pharmaceutical industry depends on worldwide coordination of clinical trials. Regulatory standards rely on shared frameworks. Safety signals are detected through international data sharing.
The WHO supports the development of harmonized guidelines for clinical research, manufacturing quality, and pharmacovigilance. These guidelines reduce duplication, lower costs, and speed patient access to new therapies.
When the US withdrew, companies lost a seat at the table where these standards are shaped. Other countries will still move forward, led by Europe or China, and US firms will then face rules they did not help design. Agencies, including the CDC and NIH, have been instructed to halt official collaboration with the WHO, including co-authoring technical papers and participating in coordinated clinical trials, which previously helped US scientists quickly test treatments in diverse populations. This will create friction in drug development, increase compliance costs, and delay product launches.
Effects on Drug Safety and Quality
The WHO estimates that one in ten medical products in low and middle-income countries is falsified or substandard. These products do not stay overseas. Global supply chains mean unsafe medicines can enter the US market through imports, online pharmacies, or contaminated raw materials.
The WHO helps countries detect and stop these products before they spread. The WHO shares alerts with national regulators, including the FDA. Leaving the WHO weakens the safety net, putting US patients at greater risk of receiving ineffective or dangerous medicines.
Pandemic Preparedness and National Security
The US government has repeatedly recognized pandemics as serious threats to the economy and defense.
The WHO coordinates pandemic preparedness plans, emergency stockpiles, and rapid response teams. It helps countries share virus samples and critical research data for vaccine development. During COVID-19, early sharing of genetic sequences allowed US companies to begin vaccine development within days. That speed saved innumerable lives. Walking away from the WHO does not make the United States more independent. It makes it more isolated at precisely when cooperation matters most.
On 23 January 2026, California became the first US state to join a WHO-coordinated international network independently. California has joined the Global Outbreak Alert and Response Network (GOARN). California accessed international expertise and data for disease monitoring, allowing the state to stay connected to global health security and utilize early warning systems.
Loss of Leadership
For decades, the United States has helped shape global health policy through the WHO. This influence helped to align global health goals with values such as integrity, transparency, and accountability. The US's leaving does not eliminate the WHO; it creates a leadership vacuum. Other nations step in and shape priorities in their stead.
Without US participation, our perspectives on data sharing, regulatory science, and ethical research lose impact. This shift affects everything from outbreak reporting to drug approval standards.
Economic Consequences for the United States
Global health stability supports global economic balance. Outbreaks play havoc with supply chains, reduce workforce productivity, and slow trade. The World Bank estimates that pandemics can cost the global economy trillions of dollars. When the US invests in global disease prevention through organizations like the WHO, it reduces the risk of expensive disruptions that negatively impact businesses and workers.
Future of Global Health
Globally, health challenges are becoming more complex. Climate change is expanding the range of infectious diseases. Antibiotic resistance is rising. New viruses continue to emerge.
No single country can manage these risks alone. The WHO remains the only organization with the reach and mandate to coordinate a global response.
Renewing trust and reforming international institutions are difficult, and abandoning them is not a solution. Active participation allowed the United States to push for transparency and efficiency from within.
Leaving the World Health Organization is a mistake with real consequences. It weakens disease surveillance, slows pharmaceutical innovation, increases safety risks, and reduces US leadership worldwide.
For patients, it means greater exposure to health threats and fewer protections. For the pharmaceutical industry, it means higher uncertainty and reduced influence. For the world, it means a less coordinated response to crises that affect everyone.
Global health cooperation is not a favor to other countries. It is an investment in safety, prosperity, and leadership. Walking away from the WHO does not make the United States stronger. It makes the world, including the US, more vulnerable.
While the current administration maintains that leaving the WHO restores accountability for US taxpayers and allows more autonomous health policy, this is actually a penny-wise, billion-dollar-foolish move, leaving the US more vulnerable to the inevitable return of transnational health threats.
Connect with Metis Consulting Services today to keep your business steady while the global stage shifts. hello@meticconsultingservices.com

Are Your Quality Systems Inspection Ready?
An excellent and sustainable quality management system is the heartbeat of pharmaceutical safety and long-term compliance. This week in the Guardrail, we examine the shift from reactive audit preparation to a proactive culture of regulatory excellence.

An excellent and sustainable quality management system is the heartbeat of pharmaceutical safety and long-term compliance. This week in the Guardrail, we examine the shift from reactive audit preparation to a proactive culture of regulatory excellence.
By Michael Bronfman, for Metis Consulting Services
January 26, 2026
Quality systems are essential for every pharmaceutical company. Quality Assurance and Quality Control ensure that drugs are developed, manufactured, and distributed safely and in compliance with regulations. Inspection readiness means more than just passing an FDA or EMA inspection; it involves building a culture of quality that supports compliance, efficiency, and public trust.
Regulators expect companies to have strong quality systems, and inspections test how well these systems work in practice. Companies that start preparing only after receiving an inspection notice run into problems that could have been avoided with earlier preparation.
Understanding Quality Systems
A quality system is a group of policies, procedures, and practices that help products consistently meet requirements. It includes document control, change management, handling deviations, corrective and preventive actions, audits, and training.
The FDA provides guidance on quality systems through regulations such as 21 CFR Parts 210 and 211 for pharmaceuticals and Part 820 for medical devices.
Information is available at https://www.fda.gov/industry.
The EMA also guides good manufacturing practice and quality system expectations at Good Manufacturing Practice | European Medicines Agency (EMA)
Understanding the regulatory framework is the first step toward being ready for inspections.
Inspection Readiness Is Continuous
Inspection readiness is not a one-time task; it is part of everyday work. Internal and external (consultants, contractors, and vendors) staff need training on procedures and are expected to understand how their work supports overall quality.
Key components of inspection readiness include:
Up-to-date documentation: Standard operating procedures, batch records, validation protocols, and training records should always be current and complete.
Traceability: Actions and decisions need documentation so any process can be followed from start to finish.
Accountability: Roles and responsibilities should be clear, and staff should be able to show they understand their tasks.
Continuous monitoring: Metrics and trends are expected to be reviewed regularly to identify potential issues before they worsen.
Conducting Self Assessments
Self-assessments, or internal audits, are one way to prepare for inspections. They help to identify gaps, verify compliance, and offer staff a chance to practice answering regulator questions.
To make self-assessments and process reviews effective, review each quality system process to ensure it complies with procedures and regulations. Audits of records include randomly selected records to check for accuracy, completeness, and timeliness, mock inspections, where a regulatory inspection is simulated, help train staff, and identify weak areas. Any issues identified during self-assessments are to be remedied through corrective and preventive actions. Taking this proactive approach reduces risk during an actual inspection.
Document Control and Data Integrity
Document control is central to every quality system. SOPs, training records, batch records, validation documents, and audit reports are always to be current, well-organized, and easy to access.
Data integrity is crucial. Regulators expect records to be accurate, complete, and protected from unauthorized changes.
FDA guidance on data integrity and compliance is available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations
Maintaining strong document control and data integrity builds trust in the quality system and helps with inspection readiness.
Training and Competency
Staff training goes beyond completing courses. Employees should understand their roles, know the procedures, and demonstrate competency.
Training programs should include:
Initial onboarding on quality system expectations
Role-specific technical training
Refresher training for updates to procedures or regulations
Records demonstrating completion and comprehension
Skilled and confident staff play a significant role in successful inspections.
Corrective and Preventive Actions
No system is perfect, so deviations and nonconformities will happen. How a company responds shows the strength of its quality system. Active CAPA programs include:
Root cause analysis: Find the real reason for the issue, not just the symptom. Effective action: Address the specific issue promptly and completely
Preventive action: Make changes to stop the problem from happening again. Confirm that the CAPA has resolved the issue and improved the process.
Inspectors look closely at CAPA records. Well-documented CAPAs demonstrate that the company is proactive and compliant with regulations.
Audit Programs
Internal and external audits are essential for ongoing improvement. Regular internal audits help find gaps and improve processes. Vendor audits ensure suppliers meet quality standards and regulatory requirements.
Audits should include documented findings, follow-up actions, and checks to confirm that changes were effective. A robust audit program shows regulators that quality is actively managed.
Managing Regulatory Inspections
When an FDA or EMA inspection takes place, being prepared makes a big difference. Key steps include:
Leadership involvement: Make sure managers are visible and know what is going on.
Document accessibility: Keep records organized so they can be found quickly.
Staff readiness: Train staff to answer questions with facts and confidence.
Issue resolution: Set up a process to document follow-up and respond to any observations. Inspectors look at both compliance and the company’s culture of quality and ongoing improvement.
Leveraging Technology
Quality Systems are encouraged to use technology to work more efficiently and track progress. Electronic quality management systems (eQMS) help with document control, CAPA tracking, training management, and audit programs. Using technology properly improves traceability, reduces errors, and makes inspection preparation easier. It also helps monitor trends and risks.
Continuous Improvement
A quality system that is always ready for inspection is continuously evolving and improving. Continuous improvement helps processes develop, closes gaps, and uses lessons from audits, assessments, and daily work.
Reviewing metrics and trends, and comparing them to industry standards, helps maintain high performance. Companies that focus on continuous improvement are better prepared for inspections and achieve better quality results.
Looking Ahead
Inspection readiness is not just a checklist. It is an ongoing commitment to quality, compliance, and patient safety. Integrating training, documentation, monitoring, and continuous improvement into daily work, companies can reduce risk and demonstrate regulatory excellence, transparency, and accountability.
Organizations that embrace these expectations and maintain strong quality systems will be able to respond confidently to inspections, protect patients, and sustain long-term success.
Ready to transform your compliance strategy? Contact Metis Consulting Servicestoday to schedule a consultation with our team of experts.
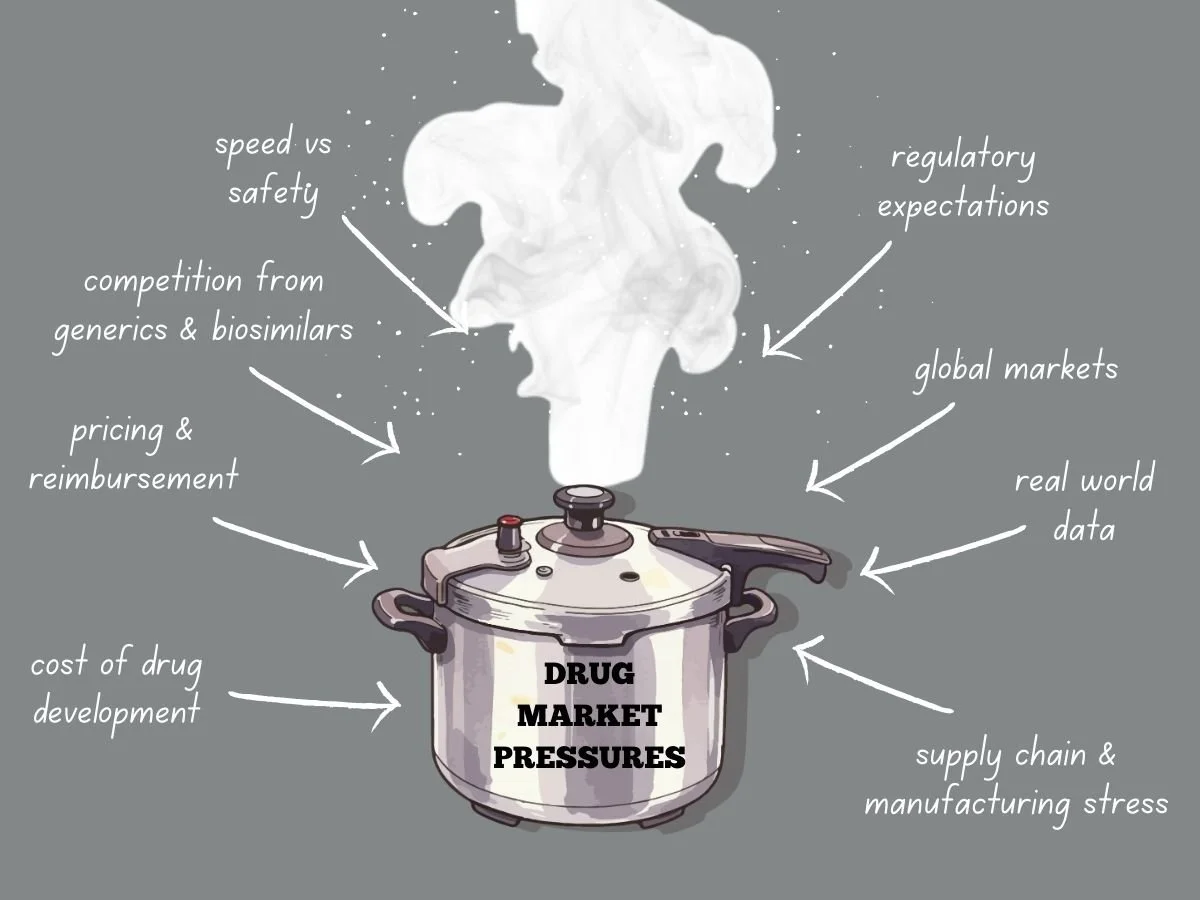
Navigating Intense Drug Market Pressures
Today’s global drug market faces more pressure than ever before. Pharmaceutical companies deal with higher development costs, tougher regulations, stricter pricing, and more competition from generics and biosimilars. Meanwhile, patients, payers, and governments want quicker access to safe and effective treatments. These factors are changing how companies plan, develop, launch, and manage products over time.
To succeed, companies need to know what causes market pressure and how it shapes their decisions. They also have to adjust their strategies while keeping quality, safety, and compliance intact.
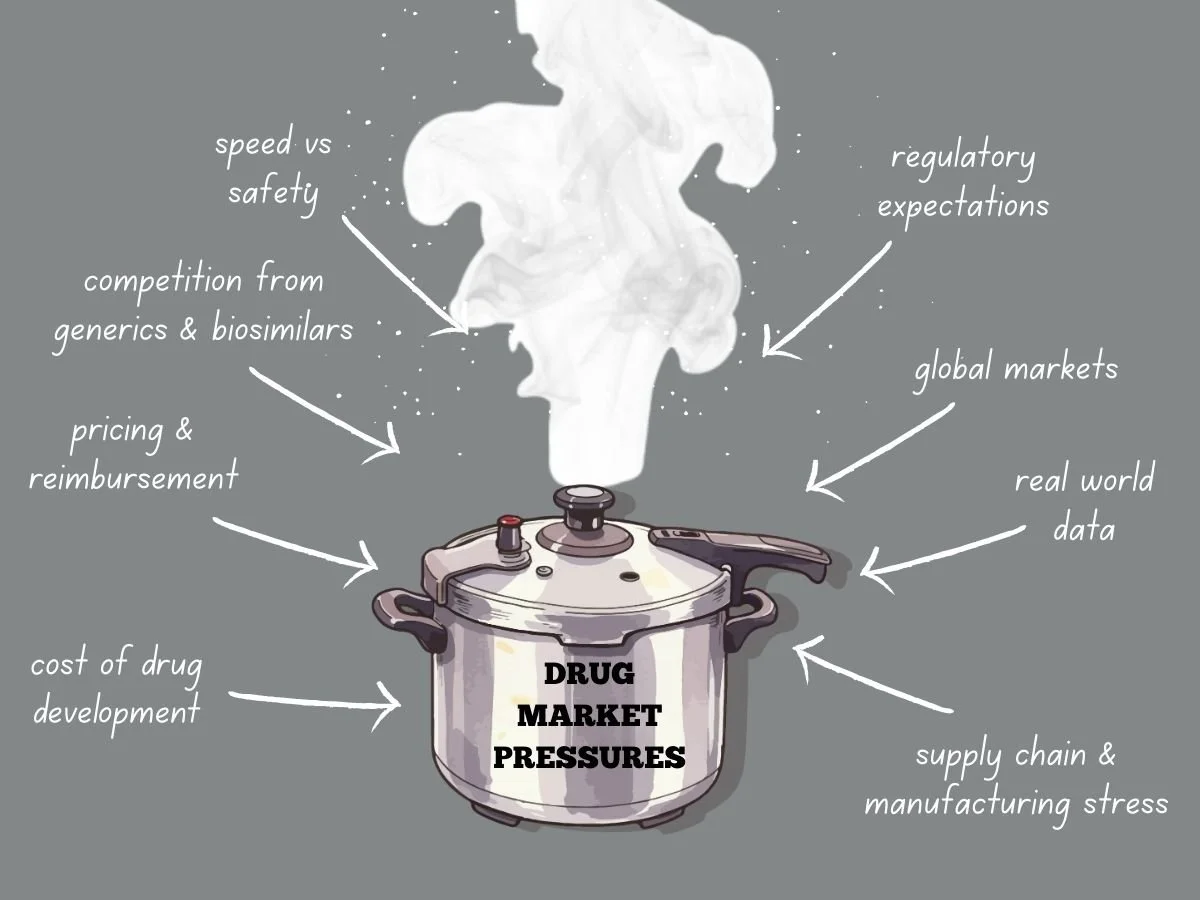
This week in the Guardrail, we explore the intensifying economic and regulatory forces reshaping the global pharmaceutical landscape. We analyze the ways industry leaders can maintain compliance and quality while navigating the high-stakes pressures of modern drug development.
By Michael Bronfman for Metis Consulting Services
January 19, 2026
Today’s global drug market faces more pressure than ever before. Pharmaceutical companies deal with higher development costs, tougher regulations, stricter pricing, and more competition from generics and biosimilars. Meanwhile, patients, payers, and governments want quicker access to safe and effective treatments. These factors are changing how companies plan, develop, launch, and manage products over time.
To succeed, companies need to know what causes market pressure and how it shapes their decisions. They also have to adjust their strategies while keeping quality, safety, and compliance intact.
The Cost Reality of Drug Development
Developing new drugs is both costly and risky. It can take billions of dollars to bring a new medicine to market, especially when you include failed attempts. Clinical trials last for years, regulatory submissions need a lot of data, and manufacturing must meet rigorous quality standards.
As development costs go up, so does market pressure. Investors want to see returns, so leaders have to focus on programs most likely to succeed. This means making tough choices about which therapies to continue and which to pause or stop.
Pressure grows when competitors work on similar products. If a company is second to market, it can lose pricing power and market share. This pushes companies to find ways to develop products faster while still following the rules.
Pricing and Reimbursement Challenges
Pricing pressure is a major challenge in today’s drug market. Governments and private payers are resisting high launch prices, and value-based pricing is becoming more common. Companies now have to clearly show clinical benefits, real-world results, and economic value.
In the United States, pricing scrutiny continues to grow through policy changes and public debate. Programs like Medicare negotiation place additional pressure on manufacturers to justify pricing decisions. The NIH National Library of Medicine is a good resource for more information at https://pmc.ncbi.nlm.nih.gov/articles/PMC11129567/
In Europe, pricing and reimbursement decisions are often made at the country level. Health technology assessments play a significant role in determining whether a product will be reimbursed and at what price. The European Medicines Agency provides regulatory approval, but market access depends on additional reviews. https://globalpricing.com/pricing-and-reimbursement-trends-in-europe-current-landscape-and-implications/
Because of these pressures, companies can not wait until after approval to plan for market access. They need to start early, thinking about evidence, comparators, and patient groups.
Competition from Generics and Biosimilars
Patent expiration is still a major source of market pressure. Once exclusivity ends, generics and biosimilars can quickly cut into revenue. Sometimes, prices fall by over 80 percent in the first year.
Companies have to plan their product life cycles early. They might consider new formulations, more uses, or combination products. Each choice needs careful regulatory planning and strong supporting data.
The US Food and Drug Administration provides guidance on generics and biosimilars, including approval pathways and exclusivity considerations at https://www.fda.gov/drugs/development-approval-process-drugs/how-drugs-are-developed-and-approved.
Companies that wait too long to plan for the loss of exclusivity often have trouble protecting their product’s value when competitors arrive.
Speed Versus Safety
Market pressure often makes teams move faster. Quicker development can help patients get treatments sooner and improve a company’s position. But moving too fast without proper controls can lead to big risks.
Rushing trials can lead to poor study design or trouble enrolling patients. If data is incomplete, it can cause regulatory delays or extra work after approval. Cutting corners in manufacturing can cause quality problems and require more inspections.
To balance speed and safety, companies need strong oversight. Clear decision rules, teamwork across departments, and early talks with regulators are key. Programs that focus on quality from the beginning handle market pressure better.
Regulatory Expectations Do Not Ease Under Pressure
One common misconception is that regulators may be more flexible when market needs are urgent. While there are faster review programs, the standards for safety, effectiveness, and quality do not change for the Fast Track and Breakthrough Therapy designation. These programs aim to speed access while maintaining standards.
The FDA provides guidance:
But it can be tricky to navigate these expectations.
Using these pathways successfully requires careful planning and ongoing communication with regulators. Market pressure is not a good reason for weak data or incomplete submissions. This adds another layer of pressure. Regulatory requirements vary by region. Clinical trial designs must support multiple agencies. Manufacturing and labeling must meet diverse standards.
If regions are not aligned, it can cause delays and extra costs. For example, a trial designed just for the US might not work in Europe or Asia. Aligning global strategy early helps avoid these problems.
The International Council for Harmonisation plays a key role in aligning technical requirements across regions. Information on these guidelines is available at https://www.ich.org/page/search-index-ich-guidelines.
Understanding and applyLearning and using these guidelines early helps companies handle global market pressure better.
Global Markets Add Complexity
Many products are developed for global markets. This adds another layer of pressure. Regulatory requirements vary by region. Clinical trial designs must support multiple agencies. Manufacturing and labeling must meet diverse standards.
Misalignment between regions can lead to delays and added costs. For example, a trial designed only to meet US requirements may fall short in Europe or Asia. Early global strategy alignment helps reduce this risk.
The International Council for Harmonisation plays a key role in aligning technical requirements across regions. Information on guidelines is available at https://www.ich.org/page/search-index-ich-guidelines
Understanding and applying these guidelines early helps companies manage global market pressure more effectively.
Manufacturing Stress
Market pressure continues after approval. Manufacturing and supply chains have their own problems. It’s hard to predict demand, especially for new products. Shortages, global issues, and quality problems can all disrupt supply. Manufacturers to maintain control and continuity of supply. Inspections focus on data integrity, process validation, and change management. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations
When under a lot of pressure, companies might try to stretch their capacity or put off investments. These choices often increase risk and can lead to regulatory problems.
The Role of Real World Evidence
As pricing and access decisions rely more on data, real-world evidence is becoming more important. Payers and regulators want to know how products work outside of controlled trials.
To collect good real-world data, companies need planning, the right systems, and oversight. They also have to meet regulatory standards for data reliability and privacy.
https://www.fda.gov/science-research
Companies that build these capabilities early are better able to handle market pressure and keep their products valuable over time.
Organizational Alignment Under Pressure
Market pressure often reveals weak spots in how organizations are set up. Gaps between clinical, regulatory, quality, and commercial teams can slow decisions. Conflicting goals can also cause teams to lose focus.
Successful organizations set shared goals and maintain open communication. They invest in training and clear processes. Leaders make it clear that compliance and quality always come first, even when deadlines are tight.
This kind of culture is essential when companies face inspections, audits, or public attention.
Looking Ahead
Drug market pressures are not going away. If anything, they will continue to intensify. Companies that treat pressure as a reason to cut corners will face setbacks. Those who use pressure as a driver for more thoughtful planning, stronger execution, and earlier collaboration will be better positioned to succeed.
Navigating this environment requires discipline, foresight, and respect for regulatory expectations. It also requires a clear focus on patients, who remain the ultimate reason these products exist.
The path from laboratory to patient has never been more complex, but you don't have to navigate these regulatory and economic hurdles alone. Contact Metis Consulting Services today. Learn how our strategic oversight and industry expertise can help your organization transform market pressure into a sustainable competitive advantage.