
Moving Beyond the Sandbox: How to Build Inspection-Ready AI in Regulated Life Sciences
For years, AI was just a tool for simple tasks like drafting emails. Those days of casual use are over. Regulators are tightening expectations, demanding strict AI governance, perfect traceability, and full integration with quality compliance systems. These rules are no longer optional best practices.
As global regulators rapidly tighten enforcement on pharmaceutical companies, this week we look at the high-stakes gap between flashy AI pilots and the rigorous, audit-ready validation required in the life sciences sector. Manufacturers must have systems in place for bulletproof governance to survive their next inspection.
By Michael Bronfman
June 30, 2026
Many pharmaceutical companies live in a frustrating middle ground. You can see it in boardrooms and IT departments everywhere. Teams enthusiastically say, “We are experimenting with artificial intelligence!” Yet when asked whether that same technology is ready for an official regulatory inspection, the room falls silent.
The gap between a cool pilot project and a fully validated, inspection-ready system is where most life sciences companies currently find themselves.
For years, teams treated artificial intelligence as a collection of non-product tools used for simple tasks. It might have been used to summarize long documents or draft email templates. But the days of casual experimentation are officially over. Regulatory bodies are tightening their expectations. They are demanding strict AI governance, perfect traceability, and complete integration with quality compliance systems. They are no longer leaving these rules to optional best practices.
Artificial intelligence systems inform labeling, product performance claims, drug dosing, patient safety, or quality decisions; they face a tough reality. The entire solution must fulfill rigorous quality, validation, and lifecycle controls. Generic pilots fail to scale because they lack the foundation required to survive a regulatory audit. Life sciences organizations must shift to purpose-built artificial intelligence that incorporates strict governance controls, unalterable audit trails, and validated results to succeed.
The Shift in Regulatory Reality
Why do generic pilots fail?
To understand this, look at how global authorities view technology. The Food and Drug Administration released major updates that signal a strong enforcement posture for advanced software used in regulated spaces. The agency treats high-risk artificial intelligence with the same seriousness as physical medical devices or critical manufacturing tools.
Traditional software validation worked well for static systems. In the past, computer systems validation followed a predictable path. A developer wrote code, a quality team tested that it did exactly what it was supposed to, and the software never changed unless an engineer manually updated it.
Artificial intelligence contradicts this old way of thinking. Advanced models are dynamic. They are built to learn, observe patterns, and develop over time. Because these systems can evolve based on the information they process, standard testing methods are inadequate. Software cannot be tested once and assumed to behave exactly the same way a year from now.
Regulators are fully aware of this challenge. They are looking closely at:
Design Controls: How the model was built, chosen, and structured.
Model Validation: Proof that the mathematical formulas produce accurate, repeatable results.
Data Authenticity: Complete certainty that the information feeding the model is clean and unaltered.
Risk Management: Clear plans for handling unexpected errors.
If an inspector walks into your facility today and sees an advanced tool helping your quality team make decisions, they will ask tough questions. They will want to know how you verify the output. They will want to see how you track changes in the system. If your only answer is that a vendor told you the tool works, you are facing a major compliance risk.
Why Generic AI Pilots Fail to Scale
It is incredibly easy to build a successful pilot project. A small team can upload historical quality data into a popular, generic large language model. Within an afternoon, the tool can review past records and suggest draft standard operating procedures or summarize corrective and preventive action reports. The team celebrates, declares the project a success, and plans to roll it out to the whole company.
Then, they meet the quality assurance department.
Generic artificial intelligence applications are built for mass productivity, not the high-stakes world of life sciences. When you try to push a basic pilot into a Good x Practice environment, the system usually falls apart for several reasons.
The Opaque Decision Process
Generic models operate like a black box. A user submits a question, and the tool provides an answer, but no one can trace the exact path the software took to reach that conclusion. In a regulated environment, an untraceable answer is a non-compliance finding waiting to happen. If you cannot prove how your software reached a conclusion about a batch failure or a clinical trial data point, you cannot use that conclusion.Missing Explicit Intended Use
Validation cannot be generic. You cannot validate an advanced tool for general office work and then use it to triage quality investigations. Every application must have a clearly defined intended use statement. This statement must outline the exact process the tool supports, who the users are, what source systems feed it data, and how the output impacts human health or product quality. Generic tools are not built to restrict themselves to a single, tightly controlled workflow.Model Drift and Information Degradation
When an advanced model interacts with new data, its internal weights can shift. Over time, the system's accuracy can degrade or change, a phenomenon termed as model drift. Generic applications do not include built-in alerts that notify you when the software becomes less accurate. Missing continuous tracking protocols, a tool that worked perfectly during a pilot in January might give flawed recommendations during an inspection in November.Poor Data Lineage and Security
Where does the information go when you type it into a generic tool? Does the vendor use your proprietary molecule data to train their public models? Many basic applications lack clear data lineage. They cannot prove who had access to the data, how it was modified, or where it is stored. This violates fundamental data validity principles that require all records to be fully traceable and secure.
The Core Pillars of True AI Governance
Transitioning from an experimental sandbox to a validated environment requires a formal governance structure. Organizations must stop treating advanced tools as simple IT upgrades and start treating them as highly regulated assets. True governance rests on five core pillars.

Pillar 1: Use Case Intake and Risk Classification
You should not give every department open access to activate advanced tools whenever they want. A mature company implements a formal intake process. Before a single line of code is written or a vendor software is purchased, the business must capture the exact purpose, ownership, and expected benefit of the tool.
The tool must be classified by risk once captured. A helpful framework divides applications into three buckets:
High Risk: Systems that support clinical decision making, patient safety, quality control inspections, or deviation management. These require absolute validation rigor, design controls, and intense testing.
Medium Risk: Tools used for operational forecasting, supply chain streamlining, or trend analysis. These require clear procedural controls and standard validation.
Low Risk: Systems used for simple productivity, basic grammar corrections, or internal meeting scheduling. These require basic security reviews but minimal validation.
By tying your compliance controls directly to the risk level, you avoid over-documenting low-risk tools while assuring high-risk applications are bulletproof.
Pillar 2: Data Controls and ALCOA+ Principles
Every piece of information used by an advanced system must comply with strict data-integrity guidelines. This means all data must be attributable, legible, contemporaneous, original, and accurate. It must also be complete, consistent, enduring, and available.
Purpose-built solutions enforce these principles by creating strong data boundaries. They restrict the software so it can only access approved, verified source systems. They block the tool from pulling random information from the public internet. Furthermore, the system must keep an immutable audit trail. Every single prompt, every generated response, and every user validation must be permanently stamped with a time, date, and user identity.
Pillar 3: Mandatory Human Review
No advanced system should operate completely on autopilot when product quality or human lives are on the line. Governance frameworks ought to mandate a qualified human reviewer to check the work.
The software acts as an assistant, not the final judge. If the tool drafts a response to a quality deviation, a trained quality professional must review the source data, verify the accuracy of the draft, and officially sign off on the record. The system must store this human verification as part of the permanent compliance history.
Pillar 4: Continuous Performance Monitoring
Because advanced software can shift over time, you need a preemptive strategy to catch errors before they reach an auditor. This involves formulating clear metrics for model exactness, sensitivity, and fault rates.
Organizations must run regular challenge tests. These tests feed the system known data sets to verify that it still produces the expected results. If the performance drops below some threshold, it must trigger an automatic alert. The tool is then taken offline or restricted until a change control process evaluates the issue and revalidates the configuration.
Pillar 5: Thorough Vendor Qualification
Most companies do not build advanced language models from scratch. They partner with IT providers or integrate specialized software into their operations. However, regulators hold you responsible for your vendors' compliance.
You must thoroughly audit your technology partners. You need to inspect their security measures, bias detection protocols, and change control processes. If a vendor pushes an unannounced software update that alters how the model reasons, your validated status could vanish instantly. You must use vendors that offer complete honesty and give you control over when updates are applied.
Applying Computer Software Assurance to Advanced Systems
The thought of validating a dynamic, learning model can terrify traditional quality assurance teams. If you try to apply old, paperwork-heavy computer systems validation methods to advanced software, you will quickly find yourself buried in endless documentation. A typical project could take eight months to complete, destroying your competitive advantage.
Fortunately, the regulatory domain has evolved. The finalized computer software assurance guidance provides a modern framework that aligns perfectly with advanced technology.
Computer software assurance flips the script on validation. Instead of spending eighty percent of your time writing exhaustive test scripts and twenty percent on critical thinking, this system tells you to spend most of your time on risk analysis and critical thinking. It allows teams to focus their testing energy on the specific functions that directly impact product quality and patient safety.
When you apply this approach to advanced technology, validation becomes manageable. Instead of testing every likely response the tool could ever generate, you focus on the workflow's configuration. You test the boundaries, the data connectors, the human review steps, and the failure modes.
Organizations that utilize this risk-based framework see massive improvements. Validation timelines can drop from several months to just a matter of weeks. This allows life sciences companies to deploy powerful, automated solutions quickly without sacrificing a single shred of regulatory compliance.
How Purpose Built Compliance Platforms Solve the Problem
Living in the gap between a pilot and a validated system is dangerous and expensive. It wastes time, frustrates engineers, and leaves your business exposed to severe regulatory penalties. The solution is to step away from generic tools and adopt systems built from the ground up for regulated environments.
This is where specialized platforms make a massive difference. For instance, companies planning to manage their complex operations turn to dedicated provider ecosystems like PSC Software. Instead of trying to force a consumer application to comply with strict global laws, organizations leverage platforms designed with compliance as a core feature.
When you look at the product offerings within the life sciences ecosystem, you can see how purpose-built tools bridge the gap. For example, managing the intense training demands of a regulated workforce requires absolute precision. Neither a manual spreadsheet nor a standard corporate training tool can withstand the pressure of an audit. Using an automated option like the ACE LMS software solution ensures that every training event, standard operating procedure update, and employee qualification is tracked inside an unalterable audit trail. This level of control perfectly aligns with the information-consistency standards required for advanced automation.
Traditional validation paperwork can slow an organization to a crawl. Converting to a digital, paperless environment using tools like ACE Validation's paperless GxP software allows teams to unify their compliance activities. With document control, corrective actions, and validation records live in a single, connected digital ecosystem, implementing and monitoring advanced technology becomes simple, enabling effortless tracking of data lineage, transparent management of system changes, and a clear, organized history for any inspector who walks through your door.
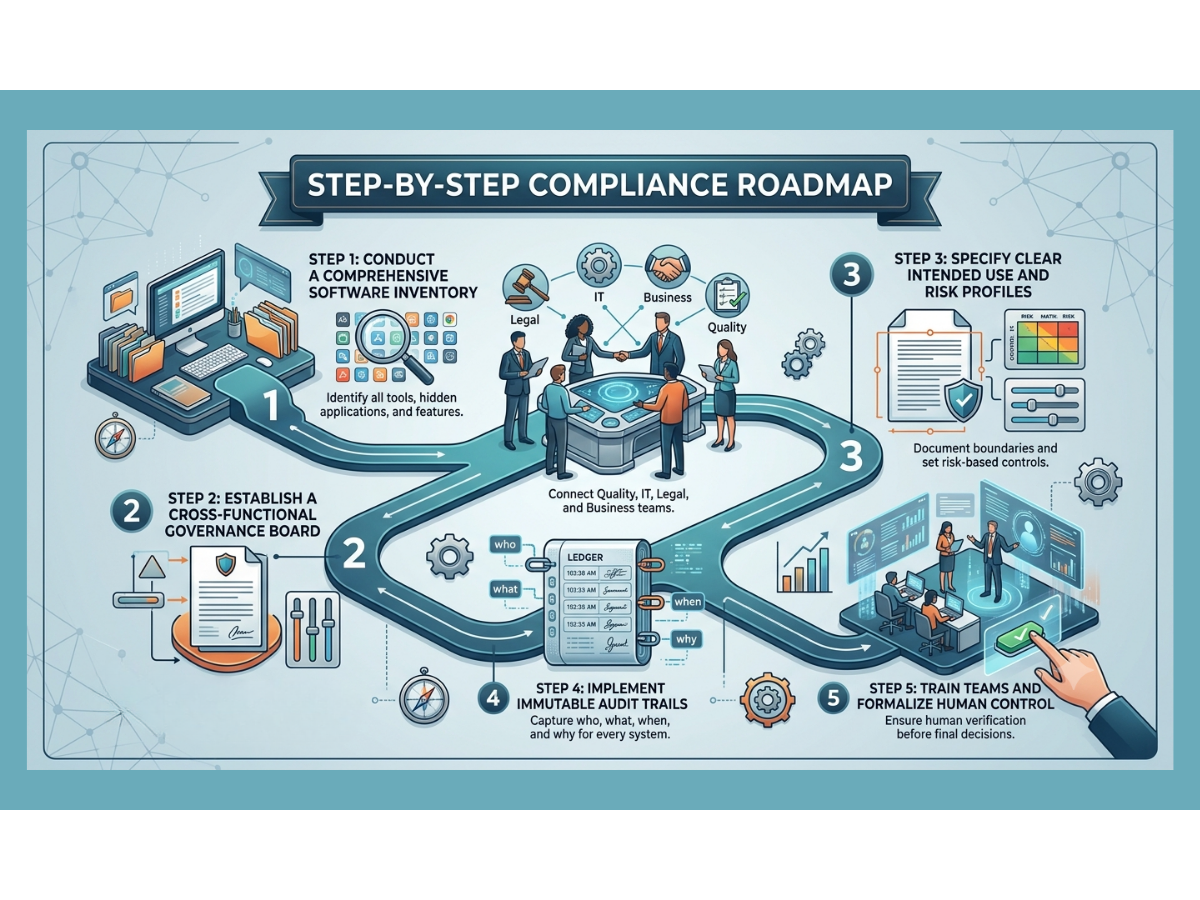
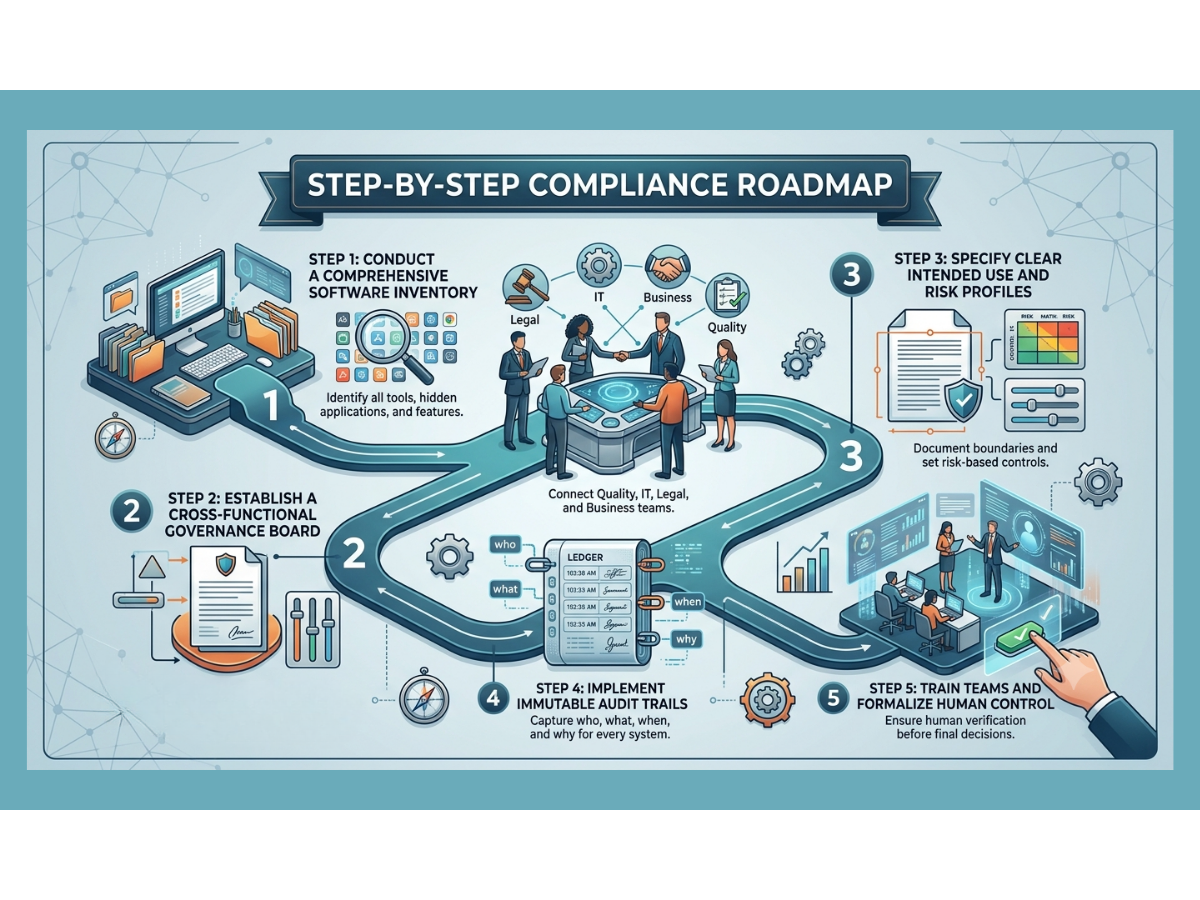
A Practical Roadmap to Inspection Readiness
If your organization wants to close the gap and build artificial intelligence systems that are truly inspection-ready, you must follow a clear, step-by-step roadmap.

Step 1: Inventory All Advanced Tools
You cannot govern what you do not know exists. Conduct a thorough audit across your business to discover every tool currently in use. Look for hidden applications where employees might be pasting company data into public websites. Document every vendor-supplied feature that claims to use smart automation.
Step 2: Create an Internal Governance Board
Bring together leaders from quality assurance, information technology, legal, and operational business units. This group will serve as the gatekeepers for all automation projects. They will review new use cases, assign risk classifications, and confirm that no project moves forward without a clear validation plan.
Step 3: Draft Clear Intended Use Statements
For every approved tool, write a detailed statement explaining exactly what the software is allowed to do and what it is strictly prohibited from doing. Document the data sources, the human review workflows, and the exact records the system will generate.
Step 4: Enforce Technical Data Controls
Work with your IT team or software vendors to confirm that every system has strong access controls, data encryption, and unalterable audit trails. Verify that the system layout prevents automatic model updates without requiring formal change control.
Step 5: Establish Continuous Monitoring Standards
Create a schedule for regular performance reviews. Define your drift thresholds and write clear standard operating procedures for what the team must do if the software shows signs of declining accuracy.
Final Thoughts
Artificial intelligence offers incredible potential for the pharmaceutical industry and can help us analyze massive data sets, spot manufacturing deviations early, and streamline heavy documentation workloads. But these benefits mean absolutely nothing if the technology cannot survive a regulatory inspection.
The era of playing around with casual pilots is over. Regulatory bodies are stepping up enforcement, and the companies that succeed will be those that treat automation with the discipline it deserves. By shifting away from generic tools and deploying purpose-built software, sound risk frameworks, and complete data lineage, you can confidently move your technology out of the sandbox and into a fully validated, inspection-ready reality.
Bridge that gap between AI innovation and regulatory reality. Contact Metis Consulting Services today. We are experts who can streamline your computer software assurance, fortify your governance structure, and ensure your technology is fully validated and completely inspection-ready.
Continuous Manufacturing and ICH Q13: Regulatory Readiness at Scale
Guided by the landmark ICH Q13 guidelines, the global pharmaceutical industry is undergoing a revolutionary shift from traditional batch manufacturing to agile, continuous production systems. Read this Week's Guard Rail to explore the revolutionary shift from traditional batch manufacturing to agile, continuous production systems.
Guided by the landmark ICH Q13 guidelines, the global pharmaceutical industry is undergoing a revolutionary shift from traditional batch manufacturing to agile, continuous production systems. Read this Week's Guard Rail to explore the revolutionary shift from traditional batch manufacturing to agile, continuous production systems.
By Michael Bronfman
June 8, 2026
The global pharmaceutical industry is undergoing a major shift in how medicines are made. For many decades, pharmaceutical factories have relied on traditional batch manufacturing. In a batch system, medicine is made in separate steps. Workers mix ingredients in a large tank, stop the machine, transfer the mixture to another station, test it for safety, and then proceed to the next step. This process takes a long time because the materials sit in wait between each phase.
Today, a newer and faster method called continuous manufacturing is changing the field. Instead of stopping and starting, continuous manufacturing moves raw materials through a single, non-stop automated system. Ingredients enter at one end of the factory pipeline, and finished tablets or liquids emerge at the other end.
This modern method affords enormous benefits, but it also creates fresh challenges for global regulators who must ensure every pill is completely safe. To help factories adopt this technology, international experts developed a set of specific rules known as the ICH Q13 guidelines. This system is helping factories around the world upgrade their machinery while keeping patient safety as the top priority.
What is Continuous Manufacturing?
To understand this industrial evolution, it helps to think about how a modern car factory works. Cars are not built by hand one at a time in separate rooms. Instead, they move down an assembly line where any part is added in a continuous, smooth flow. Continuous manufacturing applies this exact same logic to chemistry and medicine.
In a traditional batch setup, if a company wants to produce 1 million doses of a drug, it might need to run 5 separate batches. Each batch requires its own setup, cleaning schedule, and quality testing. If something goes wrong during step three of batch two, the entire batch may have to be scrapped, costing the company time and money.
Continuous manufacturing eliminates those separate steps. Machines run constantly for days or weeks at a time. Raw chemical powders are fed into the system at a precise rate and blended by automated mixers. Then they are compressed into pills and continuously coated.
This constant flow improves production. It also requires a much smaller factory footprint. A continuous manufacturing facility can often fit into a room one-third the size of a traditional batch factory, reducing energy use and building costs.
The Challenge of Process Validation and Lifecycle Management
Because continuous manufacturing runs dynamically, it cannot be monitored using old methods. In a batch system, a scientist can walk up to a large tank, scoop out a sample of powder, and take it to a lab to test its purity. In a continuous system, the material is constantly moving through pipes and tubes at high speeds. Stopping the machine to take a sample would ruin the entire production run.
This active flow elicits crucial questions concerning process validation. Process validation is the collection of data that proves a manufacturing process can reliably produce safe, high-quality medicine. Regulators require pharmaceutical companies to prove that their systems are always under control.
To achieve this control, factories use advanced tools known as Process Analytical Technology. Instead of taking physical samples, engineers place optical sensors directly inside the production pipes. These sensors use infrared light and lasers to inspect the chemical makeup of the moving powder in real time.
If the mixture deviates even slightly from the correct formula, the computer system detects the error instantly. The system can then automatically adjust the feeders' speeds or divert the flawed material to a waste bin without stopping the rest of the production line.
Managing this technology over time is known as lifecycle management. As machines age, sensors can lose accuracy, and software needs to be updated. Pharmaceutical companies must have strict plans in place to maintain, test, and calibrate these digital instruments throughout the entire lifespan of the manufacturing line.
Understanding ICH Q13 and Global Regulatory Harmony
Because different countries have their own individual health ministries, pharmaceutical companies routinely face a confusing web of rules. A factory design that is approved in the United States might face different questions from regulators in Europe or Japan. This lack of agreement can delay the release of important global medicines.
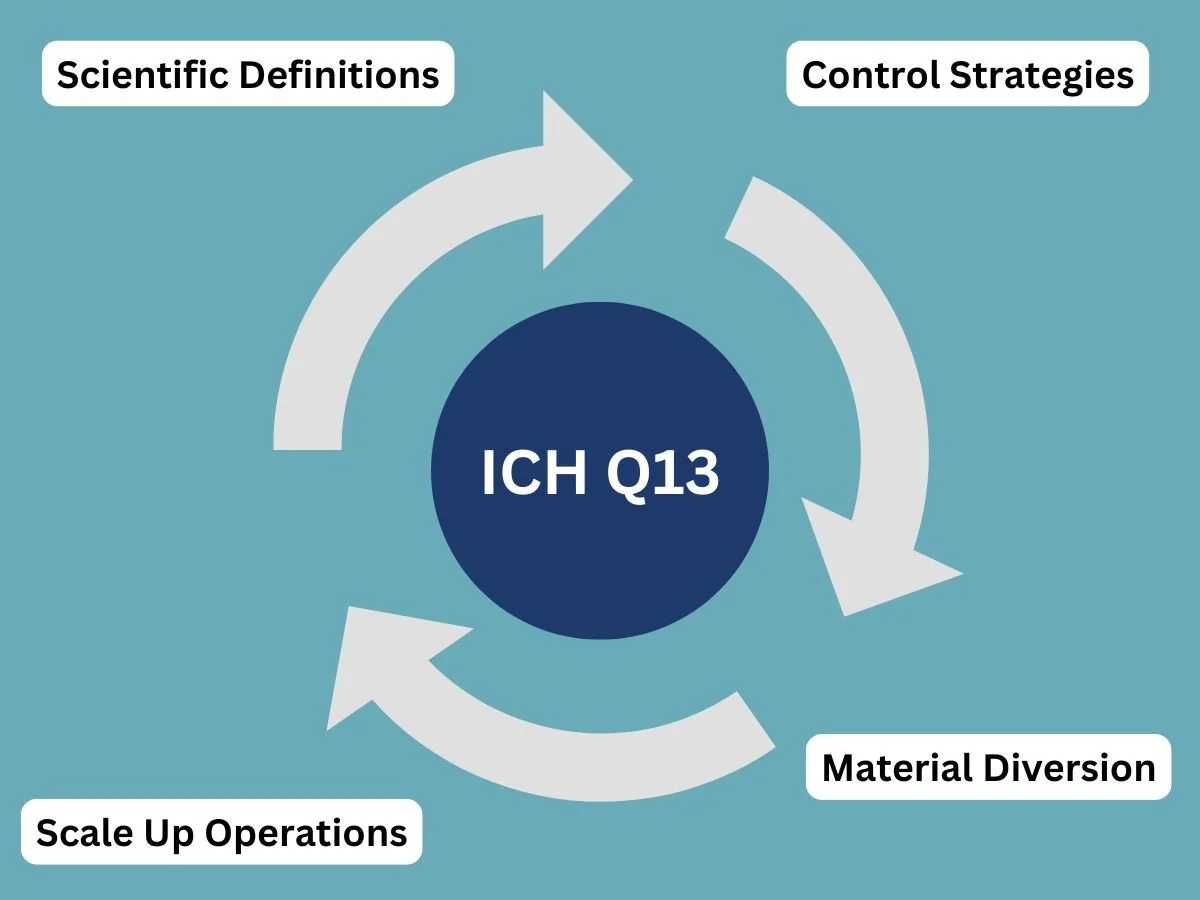
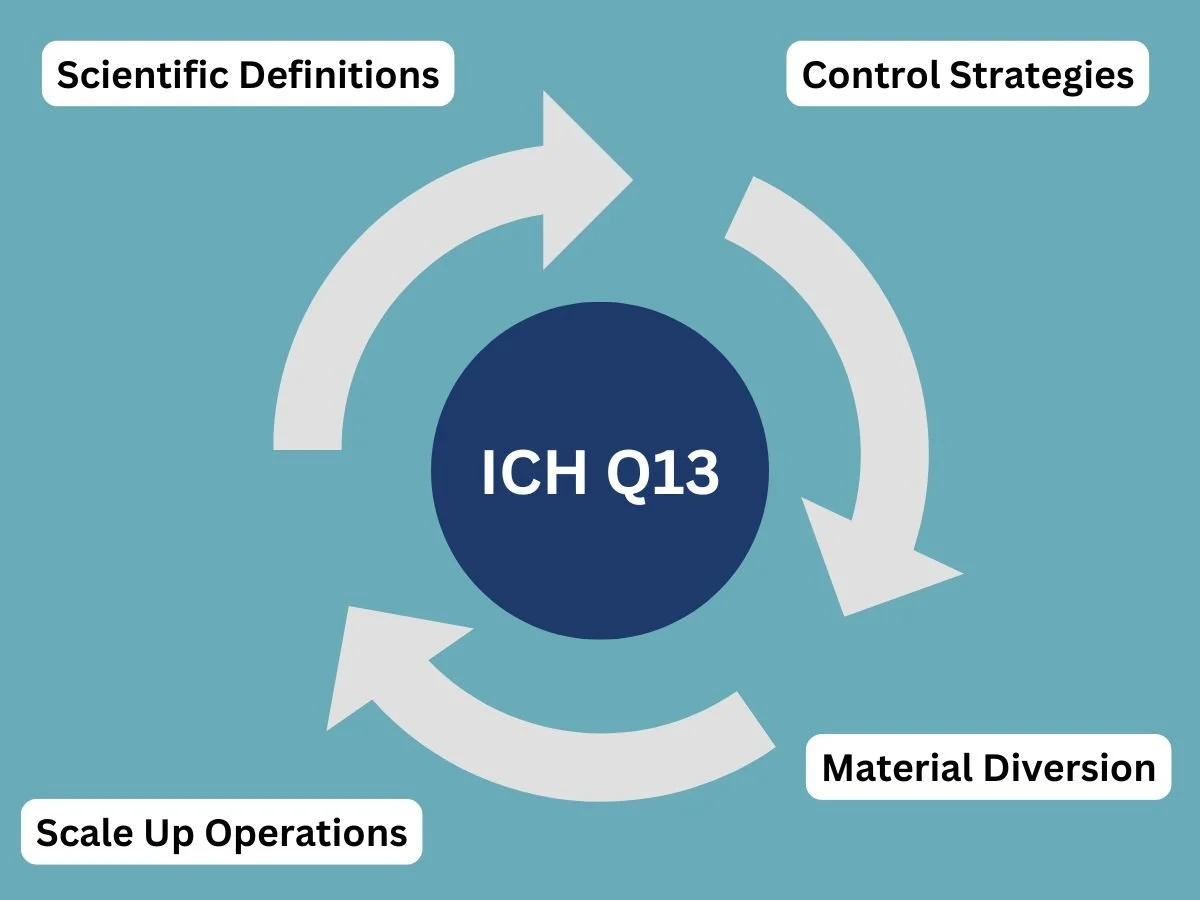
To solve this issue, the International Council for Harmonization created the ICH Q13 guideline. The goal of this document is to establish a single, internationally accepted standard for continuous manufacturing. You can read the specific technical details and formal announcements by visiting the ICH Guidance Documents page.
The ICH Q13 framework gives unambiguous instructions on how companies should handle key manufacturing concepts, including:
Scientific Definitions: Defining exactly what constitutes a batch when the material never stops flowing.
Control Strategies: Explaining how to use real-time sensors to monitor product quality.
Material Diversion: Setting rules for how and when a machine should discard substandard materials during production.
Scale Up Operations: Explaining how a company can increase production volume by simply running the machines longer, rather than building larger equipment.
By setting up these uniform rules, ICH Q13 brings global regulatory readiness to scale. It provides health inspectors with a clear checklist for reviewing these advanced facilities, thereby speeding up and making the approval process more predictable for everyone involved.
Helping Smaller Pharmaceutical Companies Innovate
In the past, only the largest global pharmaceutical corporations had the money and scientific expertise to build continuous manufacturing lines. These projects required millions of dollars in custom engineering and hundreds of hours of consultation with regulatory experts to demonstrate that the systems were safe.
The arrival of the ICH Q13 guidelines changes the landscape. Because the rules are now clearly written down and agreed upon by global authorities, the path to implementation is much easier to follow. This foreseeability makes it feasible for smaller pharmaceutical companies with less internal expertise to employ this manufacturing approach.
Instead of designing a system from scratch, smaller manufacturers can purchase pre-validated equipment that already meets international standards. They can look at the ICH Q13 document as a step-by-step blueprint for compliance. This opening of technology means that smaller companies specializing in rare diseases or generic medicines can also benefit from the efficiency, speed, and cost savings of continuous production.
Enhancing Drug Supply Chain Resilience
One of the greatest benefits of shifting to nonstop production is its contribution to the global drug supply chain. The medical world frequently faces drug shortages caused by factory delays, contaminated batches, or sudden spikes in demand during public health emergencies.
Traditional batch manufacturing is slow to react to these crises. If a hospital suddenly needs double the amount of a specific antibiotic, a batch factory has to source more raw ingredients, schedule new production slots, and run multiple separate batches over several weeks.
Continuous manufacturing solves this problem through flexibility. To scale up production in a continuous facility, you do not need to buy bigger tanks or redesign the process. You simply keep the existing machines running longer. If a machine is scheduled to run for twenty-four hours, engineers can keep it running for seventy-two hours instead.
This ability to rapidly scale production helps prevent shortages and assures that life-saving medicines remain available to patients during emergencies. For perspectives on how these supply chain improvements are being integrated into the wider medical field, you can review current industry analysis on the ISPE Continuous Manufacturing Resources Portal.
The Future of Pharmaceutical Engineering
As more factories adopt continuous manufacturing and follow ICH Q13 standards, the entire pharmaceutical domain will continue to evolve. We are already seeing the integration of fabricated intelligence along with machine learning into these automated lines. Computers can now analyze data from thousands of sensors simultaneously, predicting when a mechanical part might fail before it actually breaks down.
This high level of automation also reduces human error. Because humans do not need to manually scoop powders or transfer materials between stations, the risk of accidental contamination drops drastically. The entire process becomes cleaner, safer, and more efficient.
The transition from batch production to continuous manufacturing represents a true revolution in pharmaceutical engineering. While adjusting to these flexible validation tools entails considerable effort from both scientists plus regulators, the rewards are clear. Through international cooperation and guidelines such as ICH Q13, the pharmaceutical industry is building a more durable, scalable, and reliable system for protecting human health worldwide.
To better understand how this digital evolution affects the greater healthcare sector, we must examine how regulatory readiness shapes the commercial market. When factories adopt advanced automated systems, they do not just change their internal mechanics. They alter how quickly new therapies can reach the market.
For a closer look at how these manufacturing advancements affect actual product availability and commercial rollouts, you can track the latest pharmacy inventory updates. This connection shows that factory-floor innovation directly affects what is available on local pharmacy shelves.
Training the Next Generation of Specialists
As the industry transforms away from manual methods, the training required for pharmaceutical workers is also evolving. The modern factory floor looks more like a high-tech computer lab than a traditional chemical mixing plant.
Engineers must be fluent in data assessment, software maintenance, and mechanical engineering. They need to understand how to read complex up-to-the-minute data streams to spot microscopic variations in product density or moisture levels.
This demand for highly specialized skills has led to new partnerships between universities and industrial leaders. Educational programs are updating their chemistry and engineering courses to focus heavily on continuous processes and international regulatory frameworks.
By training students on the exact tools used in modern automated facilities, the academic world ensures that the workforce is fully prepared to operate complex systems. This educational pivot helps smaller businesses build internal expertise without hiring expensive outside consulting firms.
A Cleaner Blueprint for Global Health
Finally, the combination of advanced technology and clear international rules provides a cleaner, progressively sustainable blueprint for global public health. By limiting waste, reducing factory energy requirements, and dropping the rate of failed batches to near zero, continuous production creates a much more reliable pharmaceutical infrastructure.
When a factory runs smoothly without interruptions, manufacturing costs drop, ultimately assisting the individual patient paying for prescriptions.
The ongoing harmonization of these rules means that a breakthrough discovered in one corner of the world can be rapidly scaled up and manufactured across multiple continents using the exact same validated guidelines. This level of global readiness ensures that humanity is better prepared to address future health challenges quickly, efficiently, and in accordance with strict safety standards.
Ready to seamlessly transition your company through the complexities of ICH Q13 and the process validation, regulatory compliance, and on to the future of pharmaceutical engineering. Contact Metis Consulting Services today.

AI in Regulatory Submissions: Writing for Both Human and Machine Reviewers
This week in the Guardrail, we analyze the dual-audience reality facing modern pharmaceutical compliance. As regulatory agencies integrate automated tools to parse complex submissions, drug sponsors must adapt their documentation strategies to satisfy both algorithmic logic and human expertise.

This week in the Guardrail, we analyze the dual-audience reality facing modern pharmaceutical compliance. As regulatory agencies integrate automated tools to parse complex submissions, drug sponsors must adapt their documentation strategies to satisfy both algorithmic logic and human expertise.
By Michael Bronfman
May 25, 2026
The world of making and approving medicines is going through a massive shift. For decades, pharmaceutical companies wrote drug applications for just one audience: human scientists. Teams of medical doctors, chemists, and statisticians at agencies like the Food and Drug Administration would read thousands of pages of text to decide if a new drug was safe.
Today, that process looks very different. Pharmaceutical companies now use computer algorithms, known as Artificial Intelligence, to run clinical trials and analyze data. At the same time, the regulatory agencies themselves are starting to use computer programs to help read and sort through massive piles of application documents.
This means medical writers and drug sponsors must now write for two very different audiences at the same time. They must write for the human experts who make the final decisions, and they must write for the machine reviewers who scan the text for errors and patterns. If an application is not structured correctly for a machine to read, it could get flagged for inconsistencies before a human expert even looks at it.
To help companies navigate this change, the Food and Drug Administration released official draft guidance about using these advanced computer models in drug development. This document outlines exactly how the agency looks at data generated by computers and how companies should share that information. For more detailed context, you can read the official announcement on the FDA Press Release Page.
The Food and Drug Administration Risk Framework
The official policy from the government makes one thing very clear: not all computer applications are treated equally. The agency uses a risk-based framework to grade how much scrutiny a system needs. This framework is based on two main ideas: model influence and decision consequence.
Model influence means how much the computer output affects the final decision. If a computer makes a final choice on its own, its influence is strong. If a human expert checks the work and can override the computer, its influence is lower. Decision consequence means what could go wrong if the computer makes a mistake. If a computer error harms a patient, the consequences are high. If an error just slows down a factory machine for an hour, the consequence is low.
By looking at these two factors, the government separates computer tools into high-scrutiny systems and low-requirement systems.

High Scrutiny Systems
The highest level of official review is saved for computer systems that directly create evidence for a drug application. These are systems where a mistake could directly hurt a patient or ruin the results of a scientific study.
The government pays closest attention to these five specific areas:
Patient Stratification: Choosing which patients get to be in a clinical trial based on their genetic codes or medical histories.
Dose Optimization: Using mathematical models to calculate exactly how much medicine a patient should take to get better without getting sick from side effects.
Real World Data Analysis: Scanning millions of electronic health records from hospitals to see how a drug performs in everyday life outside of a controlled trial.
Safety Signal Detection: Watching patient data in real time to spot rare and dangerous side effects before they become a widespread public health crisis.
Endpoint Derivation: Using wearable sensors like smartwatches to measure how well a patient is moving or sleeping during a clinical trial.
If a company uses a computer for any of these tasks, it must prove the system is incredibly reliable. They must show how the model was trained, what data it used, and how it avoids bias.
Low-Requirement Systems
On the other side of the coin, some computer uses do not impact patient safety at all. If a company uses a computer tool to format a document, check page numbers, or organize internal administrative tasks, the government does not need to see piles of validation data. These internal operations face proportionally lower requirements because a mistake by the computer will not change the scientific conclusions of the drug trial.
Understanding the Double Audience
Because regulatory agencies are now using advanced software to help manage incoming applications, drug sponsors must realize they are writing for a double audience. The text must satisfy both the human brain and the computer algorithm.
To see how these two audiences read differently, look at this comparison:
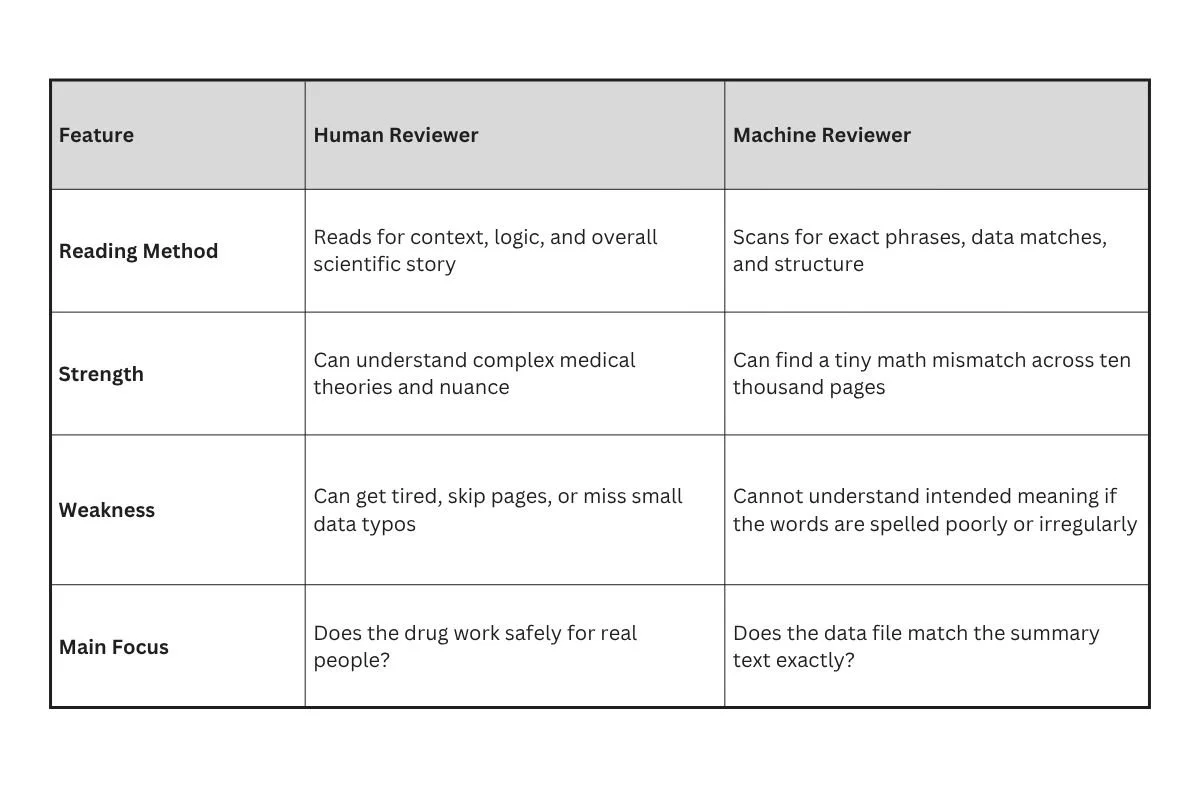
When a human reads a drug application, they want a clear narrative. They want to understand the journey of the drug from the laboratory to the clinic. They care about scientific logic.
A machine reviewer does not care about stories. It treats the document like a database. It looks at the tables, the labels, and the numbers to make sure everything adds up perfectly. If the summary on page five says fifty patients had a headache, but the raw data table on page nine hundred says forty-nine patients had a headache, the machine will flag that instantly. A human might miss that small slip, but a machine never will.
Writing for the Machine Reviewer
Writing for a computer means changing how you present text. Computers like clean organization, predictable patterns, and explicit language. If you write with vague words, the software can get confused and flag your document as a risk.
Structure and Predictability
The best way to help a machine reviewer is to use standard templates. Regulatory documents should follow strict structural rules. Use clear, standardized headings for every section. Do not try to be creative with section titles. If the standard title is Clinical Efficacy, do not change it to How Well the Drug Worked. The computer looks for specific keywords to map the document, and changing those keywords breaks the map.
Data Consistency and Labels
Every data point must look identical throughout the entire file. If you refer to a drug concentration as ten milligrams on one page, do not write it as 10mg on the next page. Choose one format and stick to it.
Also, make sure that every chart and table has clear, descriptive labels that use text instead of scanned images. Machine reviewers read text characters, not picture pixels. If you paste a picture of a table into your document, the computer sees a blank space and misses all the important data inside it.
Front Loading for Clarity
Machines are built to look for core conclusions early. Put your main findings, safety summaries, and essential data points right at the front of your sections. Do not hide your main message under paragraphs of introductory fluff. Front loading your clarity helps the computer categorize your document correctly on its very first pass.
Writing for the Human Reviewer
While you must make your document easy for a computer to analyze, you cannot forget the human being who must sign the final approval paper. Humans need context, clear explanations, and a believable scientific argument.
Explaining the Why
A machine can show that a number changed, but only a human can explain why it changed. If a clinical trial had a sudden drop in patient attendance during month four, a machine might flag it as a data error.
The human writer needs to explain the context:
"Patient attendance dropped in month four due to a historic blizzard that closed three major clinical trial sites for two weeks, but patients resumed their regular visits as soon as the roads cleared."
This explanation satisfies the human reviewer and prevents them from rejecting the data.
Keeping the Story Alive
A good regulatory submission tells a story of safety and success. The human writer must connect the dots between different pieces of research. Show how the animal studies predicted the human results, and show how the human results match the goals of the project. Use active, plain verbs to explain what the scientists did. Avoid overly dense language that puts the reader to sleep. A tired reviewer is a frustrated reviewer.
Conducting an Internal Review
Before you click the submit button to send your drug application to the government, your team should perform a complete internal review. This means testing your document against your own software tools to see what a machine reviewer will find.
Step One: The Automated Consistency Check
Run your completed document through text-matching software. This program should look for every number, percent, and statistical value to make sure they match perfectly across all chapters. If the software finds a conflict, fix it immediately. You want to find these errors yourself rather than letting the government find them first.
Step Two: The Structure Audit
Verify that every hyperlink works and leads to the correct appendix. Check that your document map functions properly and that all headings match the standard table of contents. If a machine cannot navigate your document links, it may automatically reject the file.
Step Three: The Human Readability Pass
Have a scientist who did not write the document read it for flow and clarity. Ask them if the arguments make sense and if the explanations are easy to find. This step ensures that once your document passes the computer gates, it will please the human experts.
The Path Forward for Drug Developers
The use of computer intelligence in regulatory submissions is not a temporary trend. It is the permanent future of medicine. Drug companies that learn how to write for both humans and machines will get their medicines approved much faster. Those who stick to old ways of writing will face constant delays, data flags, and rejection notices.
To keep up with these changes, companies should train their medical writers in basic data science principles. Writers do not need to learn how to code, but they do need to understand how computers read and sort information. By focusing on predictability, exact data matches, and clear summaries, you can create a document that satisfies the cold logic of a machine and the deep wisdom of a human scientist.
To learn more about how the government views these new digital tools, you can review the comprehensive resources provided by theFDA Artificial Intelligence Development Page. Staying informed about these official updates is the best way to ensure your future submissions are successful.