

European Union Pharmaceutical Industry Package
In December 2025, negotiators reached a historic political agreement on a major overhaul of drug laws known as the EU pharma package. As these massive new rules roll out across Europe, pharmaceutical companies face a complicated double reality. They must adapt to a unified European market while simultaneously managing messy, localized differences from country-specific ethics boards and regional testing requirements.

This week in the Guardrail, we dive into the sweeping changes brought by the landmark EU pharma package agreement and its impact on global drug development. Learn how balancing unified European regulations with intricate local hurdles is redefining pharmaceutical strategy as we advance through 2026.
By Michael Bronfman
July 13, 2026
Imagine trying to launch a new smartphone, but every single city you sell it in has a completely different rule about how the battery must work, how the screen must be tested, and what information needs to be printed on the box. You would spend all your time filling out paperwork instead of actually building great technology.
This exact challenge happens every day in the medical world. It is known as global regulatory divergence, which is a fancy way of saying that different countries have vastly different laws for approving and monitoring new life-saving medicines.
For decades, international organizations have tried to create global regulatory harmonization, which means getting health authorities across the globe to agree on a single, shared rulebook. When rules match, safe medicines can reach sick patients much faster.
However, achieving this balance is an ongoing tug-of-war. A massive example of this shift is happening right now in Europe. In December 2025, negotiators reached a historic political agreement on a major overhaul of drug laws known as the EU pharma package. As these massive new rules roll out across Europe, pharmaceutical companies face a complicated double reality. They must adapt to a unified European market while simultaneously managing messy, localized differences from country-specific ethics boards and regional testing requirements.
The EU Pharma Package: A Balanced Approach to Innovation
The newly agreed-upon European rules represent the most significant update to continental drug laws in over twenty years. The policy balances two urgent goals: getting critical treatments to patients much faster and providing strong, predictable rewards to companies that invest billions of dollars into medical discoveries. The continent is modernizing the evaluation of medicines throughout the entire life cycle of a drug.
A major feature of this update is its handling of regulatory data protection. When a pharmaceutical company invents a brand-new medicine, they receive a specific window of time during which competitors cannot copy their laboratory data to make cheap, generic versions. The new framework modernizes this timeline by offering extended data and market protection terms under specific conditions.
If a company creates a drug that treats a totally unaddressed disease, or if they launch the drug across every single European member state simultaneously, they can earn extra months of exclusive market control. This clever setup uses market access as a reward, encouraging companies to launch their newest treatments in smaller or less wealthy nations instead of just focusing on the biggest, richest markets.To explore the official steps and structural timelines of this historic legal rewrite, visit the European Medicines Agency Legislation Reform Portal.
Managing Supply Obligations and Preventing Shortages
Another major pillar of the new European rules targets a problem that has plagued hospitals for years: medicine shortages. We have all seen news stories about pharmacies running out of basic antibiotics or critical cancer therapies. The updated European rules address this by creating strict, legally binding rules for managing supply obligations.
Pharmaceutical companies are now required to monitor their supply chains with extreme precision. If a factory faces a production delay, the company must alert health authorities months in advance. They must also maintain emergency reserve stocks of critical medicines.
While these rules protect public health, they add a heavy layer of administrative work for drug developers. Companies can no longer just focus on science; they must build highly advanced data-tracking systems to prove to European regulators that their supply chains are secure and resilient.
Local Divergence: The Secret Layer of Review Delays
Even though the master rules are becoming unified across the European continent, real-world drug development often trips over a quieter, localized hurdle. A major central agency like the European Medicines Agency might give a new drug a green light, but a company cannot automatically hand that medicine to a patient.
Before any clinical trial can begin or any new therapy can be integrated into a local hospital, the project must pass through country-specific ethics committees. These local boards review studies to ensure human participants are treated fairly and that local cultural and legal boundaries are respected.

This is where true divergence shows its face. An ethics board in Germany might have a completely different requirement for patient data privacy compared to an ethics committee in Spain or Italy. One country might demand that patient consent forms be written in a highly specific linguistic style, while another might require extra blood tracking tests that the original master protocol did not include.
These local variations create a hidden labyrinth of review layers. If a pharmaceutical firm does not anticipate these regional differences, they will face sudden, cascading delays that can stall a global drug launch for months.
Navigating Local Intelligence and Change Management
Because local rules can vary so wildly, pharmaceutical companies are shifting how they plan their global operations. They are relying heavily on early local regulatory intelligence. This means that years before a drug is even ready for final approval, experts are on the ground in individual countries tracking the specific, unwritten preferences and shifting habits of regional boards.
To see how these international compliance standards are managed digitally across different regions, you can check the systems designed by PSC Software Compliance Systems. This type of specialized data tracking helps companies log changing regional requirements in real time, ensuring that an update made to a study protocol in one nation does not accidentally break a compliance rule in another.
Having local intelligence is only half the battle. A company must also possess effective change management systems. When an ethics committee in one country demands a sudden modification to a clinical trial or a manufacturing process, that change can trigger a massive chain reaction.
The company must update its laboratory records, notify packaging plants, alter digital labels, and inform clinical investigators worldwide. Without a rock solid change management strategy, a single localized request can cause a total operational breakdown across the entire global pipeline.
The Landscape of Drug Discovery Trends
As we advance through 2026, these regulatory shifts are fundamentally altering modern drug discovery trends. In the past, scientists would discover a molecule, test it in an isolated lab, and hand it off to a legal team to deal with compliance later. That old, disconnected model is no longer sustainable.
Today, regulatory strategy is baked directly into the earliest phases of scientific research. If a research team knows that Europe offers massive incentives for treating unmet medical needs, they will deliberately steer their laboratory focus toward rare, untreatable conditions.
Similarly, because supply chain tracking is now a strict legal requirement, discovery teams are choosing chemical ingredients that are easy and reliable to source globally, avoiding rare components that might trigger a supply chain alert later. The legal framework is actively shaping the molecular science of modern medicine.
Harmonization and Divergence in a Connected World
The ultimate goal for global health remains a world where a safe medicine can be developed once and deployed everywhere without unnecessary friction. Steps like the European pharma package prove that large-scale harmonization is possible, turning a collection of independent countries into a predictable, unified marketplace for healthcare innovation.
Yet, the persistence of local ethics boards, regional legal updates, and country-specific demands reminds us that complete uniformity is an illusion. True success in modern pharmaceuticals requires a delicate balance of macro and micro strategies.
Companies must design their master plans to align with sweeping global agreements, while keeping their operational teams agile enough to respect and adapt to local differences. By mastering this complex balancing act, the medical community can ensure that brilliant scientific discoveries do not get trapped in bureaucratic paperwork, but instead find their way into the hands of the patients who need them most.
Don't let the complex labyrinth of localized compliance, supply chain obligations, and shifting ethics board requirements stall your global drug launch. Contact Metis Consulting Services today

Understanding the New ICH-M14 Safety Guidelines
Today, the pharmaceutical industry is moving toward a massive transformation. A historic change occurred when a global organization called the International Council for Harmonization officially adopted a new framework named the ICH M14 guideline. This framework changes the rulebook for drug safety.

In the Guardrail this week: Explore the pharmaceutical industry's massive paradigm shift from isolated clinical trials to real-world data.
By Michael Bronfman
July 6, 2026
Imagine walking into a doctor's office, picking up a prescription, and knowing that the medicine you are about to take is being monitored by a global web of digital information. For decades, the gold standard for testing medicines has been the traditional clinical trial. In those trials, scientists test a new drug on a small, highly selected group of people under perfect conditions. This process works well, but it does not always show how a drug performs in the messy, complicated real world, where people forget to take pills, have multiple health conditions, or mix different prescriptions.
Today, the pharmaceutical industry is moving toward a massive transformation. Medical tracking is shifting from isolated labs to everyday life, powered by real-world data. This data includes everything from electronic health records kept by hospitals to insurance claims and tracking apps. When researchers analyze this everyday data, they generate real-world evidence that provides a clearer picture of how drugs affect diverse populations.
A historic change occurred when the global organization, the International Council for Harmonization, officially adopted a new framework, the ICH M14 guideline. This framework changes the rulebook for drug safety. It elevates everyday medical information into a form of regulatory currency, meaning health authorities now treat this tracking data with the same respect as traditional laboratory research. This shift changes the future of medicine, creating new ways to develop treatments while presenting major challenges regarding data privacy and access.
Understanding the New Safety Guidelines
To understand why this is such a major shift, it helps to look at how medicine tracking used to work across different borders. In the past, if a pharmaceutical company wanted to demonstrate that a drug was safe in both the United States and Europe, it often had to run separate observational studies in each region. Different countries had different rules about what made data reliable, how statistical math should be done, and how reports should be written. This fragmentation slowed down safety checks and made life-saving drugs take longer to reach patients who needed them.
The new global standard solves this problem. This agreement brings the world's major health authorities onto the same page, including the United States Food and Drug Administration and the European Medicines Agency. The official policy is detailed directly at ich.org, which explains how countries are unifying their rules. By creating a single set of expectations, a study built in one country can now be accepted by regulators worldwide.
This framework specifically targets non-interventional studies. These are research projects where scientists do not give patients a new drug or alter their treatment. Instead, researchers simply look backward or watch from a distance, studying how a medicine behaves as people use it naturally. Because these studies rely on information that already exists in hospital databases or pharmacy logs, having a strict global standard ensures nobody cuts corners or manipulates the findings.
Why Pre-Specification is the Key to Trust
One of the biggest concerns with observational research is a practice known as data dredging or cherry picking. Imagine a researcher looking through millions of patient records without a clear plan. If they look long enough, they might find a random pattern that makes a drug look incredibly safe or dangerously harmful, even if that pattern is just a coincidence.
The new framework eliminates this risk by mandating protocol pre-specification. This means that before scientists even look at the patient data, they must write down an exact plan detailing what they are looking for, how they will define a side effect, and how they will handle their math. This plan is locked in place so researchers cannot change their questions halfway through the study to get the results they want.
This approach builds public trust and satisfies strict regulators. When pharmaceutical companies submit their findings, they must prove they followed their blueprint perfectly. This level of planning turns casual healthcare records into high-quality scientific proof that can justify keeping a drug on the market or expanding its use to new groups of patients, such as children or elderly populations who are often left out of original clinical trials.

The Elements of Modern Evidence Packages
As these strict standards take hold, the way pharmaceutical companies present their discoveries is changing. The industry is moving away from simple stacks of paper toward dynamic evidence packages. These modern files combine multiple streams of information into a single master profile for a medicine.
A modern evidence package brings together three main components:
Clinical Trial Data: The traditional, highly controlled laboratory tests that prove a drug can work under ideal circumstances.
Real World Evidence: The continuous tracking of millions of patients using the medication in everyday situations to see how it performs across different ethnicities, ages, and lifestyles.
Digital Biomarkers: Measurable data collected from smartwatches, continuous glucose monitors, and wearable fitness trackers that show how a patient responds to a drug hour by hour in real time.
When these three streams merge, regulators get a rich picture of a drug's true impact. For example, a heart medication might show perfect numbers in a traditional lab trial. However, the wearable smart sensors might show that patients feel dizzy for an hour right after taking it, while hospital records might show fewer long term heart attacks. This complete view helps doctors make better decisions and helps pharmaceutical companies spot risks or secondary benefits much faster than before.
The Barriers of Real World Information
While this data-rich future sounds amazing, it faces significant real-world roadblocks. The first major hurdle is that most healthcare data was never designed for scientific research. When a doctor types notes into an electronic medical record or a hospital submits an insurance claim, their primary goal is to treat the patient and get paid, not to run a clinical study.
This reality creates massive problems with missing or messy data. A doctor might forget to record how much a patient smokes, or a hospital might change the way they code a specific disease mid-year. If researchers try to run high-level statistical analyses on broken information, they will get inaccurate results. Turning raw hospital paperwork into fit-for-use data requires an immense amount of cleaning, sorting, and verifying, which takes time and expensive technology.
The second massive obstacle involves access restrictions and data silos. Medical information is highly personal, and laws like the Health Insurance Portability and Accountability Act in the United States protect patient confidentiality. Because of these vital privacy laws, hospital systems, insurance firms, and tech giants often keep their data locked tightly inside their own networks.
Breaking down these walls without compromising patient privacy is incredibly difficult. If a pharmaceutical company cannot access a wide enough pool of data, their study will not represent the whole population. This leaves them unable to meet the strict global standards required by modern regulators.
The Role of Pharmacoepidemiology in Public Health
The science driving this entire movement is pharmacoepidemiology, the study of the uses and effects of drugs in large populations. This field acts as an early warning system for public health. When a new medicine hits the market, it might have been tested on only a few thousand individuals. If a dangerous side effect occurs in only one out of every fifty thousand people, a traditional clinical trial will likely miss it entirely.
Through large-scale tracking, scientists can monitor millions of prescriptions simultaneously. If a sudden spike in kidney issues appears among patients taking a specific arthritis medication, researchers can spot the trend within weeks instead of years. The new standard gives these scientists a clearer roadmap for designing these studies, ensuring their alerts are based on rigorous math rather than false alarms.
For an in-depth look at how these safety networks operate, the European Network of Centers for Pharmacoepidemiology and Pharmacovigilance provides resources showing how global networks cooperate to trace medicine safety across whole continents. This coordinated surveillance saves lives by ensuring that when a drug risk is discovered anywhere in the world, safety warnings are updated immediately everywhere.
How Health Authorities are Implementing the Standard
As we move through 2026, nations are actively weaving this framework into their daily operations. The transition requires regulatory agencies to rewrite their local playbooks to support the shared global model.
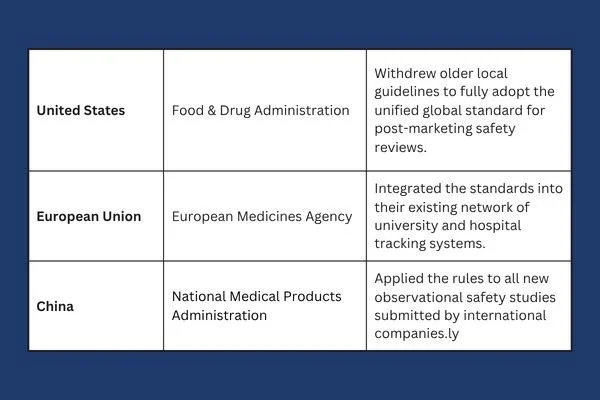
This level of cooperation is rare in international trade, but it shows how vital real world tracking has become. To explore the exact implementation details and view the official updates for American medicine, you can read the documentation here. This page shows how older local frameworks are being replaced to make room for this new way of reviewing drug safety.
The Future of Drug Discovery Trends
Looking ahead, this standard will alter more than just post market safety tracking; it will transform how drugs are discovered and developed from the very beginning. Historically, bringing a single drug to market has taken over a decade and cost billions of dollars. Much of that time was spent waiting for traditional trial results.
By using everyday tracking data as a recognized regulatory asset, companies can now design smarter trials. Scientists can study existing patient databases to discover which specific genetic groups respond best to an experimental treatment before they ever recruit a human volunteer. This approach reduces trial sizes, cuts costs, and protects human participants from taking experimental therapies that are unlikely to help their specific condition.
Furthermore, this framework opens the door for adaptive trials. In these modern studies, researchers can modify an ongoing trial based on incoming real world information, adding new patient groups or adjusting dosages safely with the blessing of regulators. The boundary between the research lab and the everyday clinic is fading away, creating a continuous loop of medical learning.
Balancing Innovation with Ethical Protection
As data becomes the lifeblood of modern medicine, the pharmaceutical industry must handle its new power with caution. Collecting digital footprints from hospital visits, insurance bills, and wrist sensors requires a steadfast commitment to patient ethics. People must be certain that their personal health struggles will never be sold, leaked, or used against them by employers or commercial firms.
The new global standard addresses this by demanding high levels of data transparency and strict data management rules. Companies must explicitly detail how they protect patient identities, strip out personal tracking markers, and secure their databases from cyber threats. Innovation is only valuable if patients feel secure using the systems that monitor them.
The transition to treating real-world information as an official currency marks a massive step forward for human health. It acknowledges that clinical trials, while vital, are just the opening chapter of a drug's true story. By turning everyday experiences into reliable science, the global medical community ensures that the medicines of tomorrow will be safer, more effective, and customized for the real world we all live in.
If your organization needs to convert messy healthcare data into high-quality, audit-ready scientific evidence that meets major global health authorities' requirements, we can help. Contact Metis Consulting Services today
ICH Q12: Post-Approval Change Management for Pharmaceutical Product Lifecycle Management
The formalized roll-out of ICH Q12 mechanisms introduces an organized approach to identifying and managing regulatory commitments. This framework allows manufacturers to execute routine modifications under the oversight of their internal Pharmaceutical Quality System (PQS), reducing the burden of prior-approval regulatory filings.
This week in the Guardrail, we break down the practical mechanics of the ICH Q12 framework. Tools like Established Conditions and Post-Approval Change Management Procedures can streamline regulatory paths and protect global supply chains.
By Michael Bronfman
May 29, 2026
The global pharmaceutical regulatory framework is transitioning from a rigid, reactive paradigm to an anticipatory, science and risk-based lifecycle management model. Central to this transformation remains the international implementation of the International Council for Harmonization (ICH) Q12 guideline.
Historically, post-approval changes (PACs) to chemistry, manufacturing, and controls (CMC) required extensive, multi-jurisdictional regulatory reviews. These extended processes frequently delayed the introduction of manufacturing innovations, equipment upgrades, and site transfers.
The formalized roll-out of ICH Q12 mechanisms introduces an organized approach to identifying and managing regulatory commitments. This framework allows manufacturers to execute routine modifications under the oversight of their internal Pharmaceutical Quality System (PQS), reducing the burden of prior-approval regulatory filings.
Evolving Jurisdictional Implementation Boundaries
Global regulatory bodies are adopting the tools and enablers outlined in ICH Q12 at varying paces and within specific product domains.
Health Canada Strategy
Health Canada has introduced updates to its regulatory infrastructure, denoting a step-wise integration of the ICH Q12 framework. The Biologic and Radiopharmaceutical Drugs Directorate (BRDD) updated its Health Canada Guidance on Post-Notice of Compliance Changes Framework to establish the operational boundaries for these tools.
Initial implementation focuses exclusively on Post-Approval Change Management Procedures (PACMPs) for products regulated by the BRDD, including biologics and Schedule C drugs. Under this system, the submission of qualifying PACMPs will be formally accepted following a 90-day transition period ending August 13, 2026.
Notably, Established Conditions (ECs) for all product classes and PACMPs for applications outside the BRDD fall outside the initial scope. Broader integration by the Pharmaceutical Drugs Directorate (PDD) is scheduled for subsequent phases, with particular timelines expected later in the year.
Global Agency Status
The United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) provide precedents for these tools across chemical entities and therapeutic biologics. These agencies accept regulatory submissions containing explicitly defined ECs and PACMPs, provided the manufacturer demonstrates an advanced PQS during routine facility inspections.
The differences in implementation speed underscore the need for multinational pharmaceutical operations to design global change strategies that navigate different regulatory requirements.
Functional Mechanics of ICH Q12 Regulatory Tools
The practical value of ICH Q12 relies on two interconnected instruments: Established Conditions and Post-Approval Change Management Guidelines. These tools shift the focus of regulatory dossiers from arbitrary data elements to critical-to-quality variables.
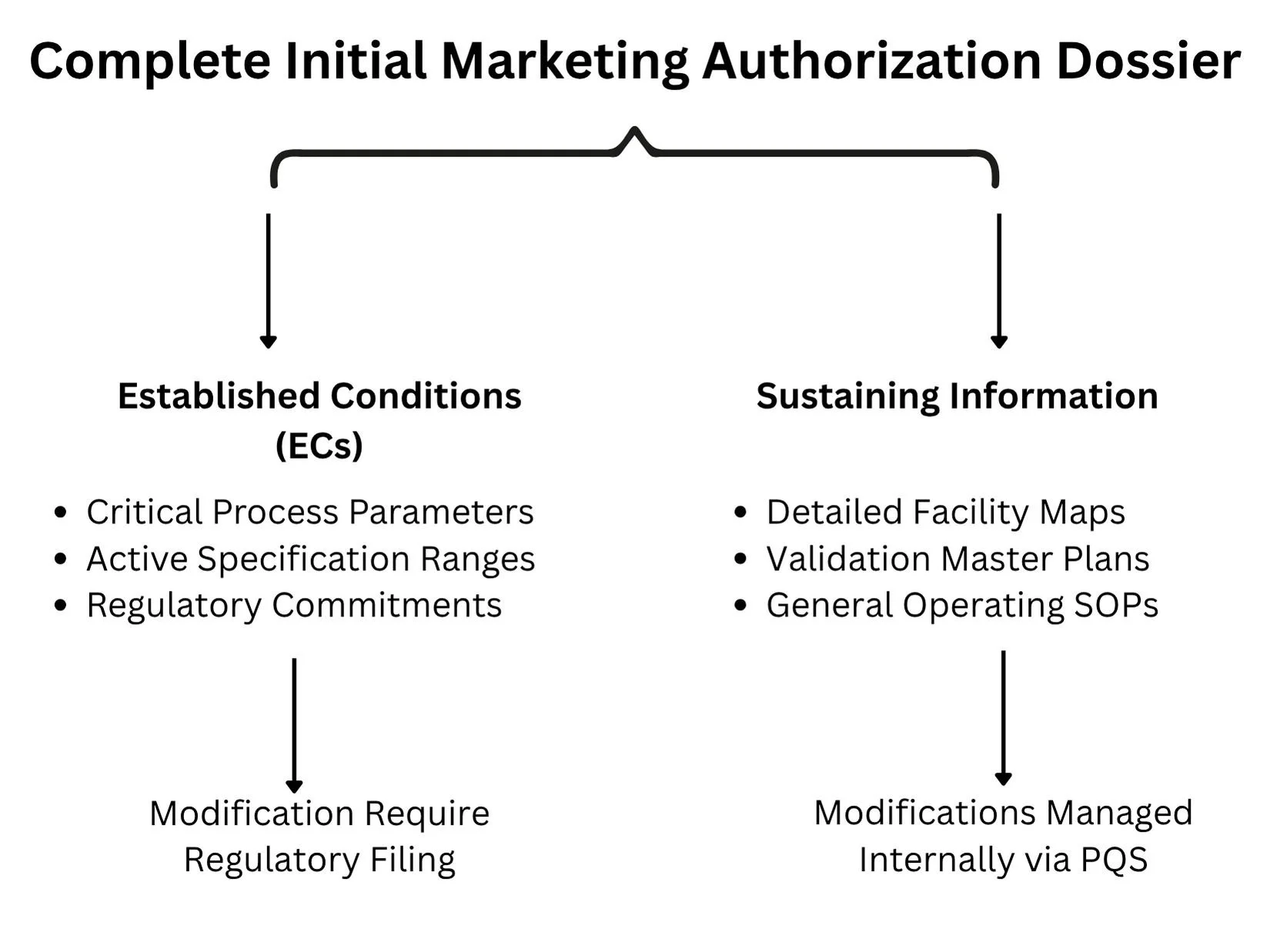
Delineating Established Conditions from Sustaining Information
A longstanding challenge in lifecycle management has been the lack of a clear distinction between legally binding regulatory commitments and purely illustrative descriptive text in the Common Technical Document (CTD). ICH Q12 tackles this by separating information into Established Conditions and Sustaining Information.
Established Conditions (ECs): Defined as the specific elements of a manufacture or control strategy necessary to ensure product safety, identity, strength, purity, or potency. Any modification to an approved EC constitutes a regulatory change that must be reported to the oversight agency. Examples include critical process parameters (CPPs), critical quality attributes (CQAs), acceptance criteria for raw materials, and active operational dimensions of specialized purification columns.
Sustaining Information: Encompasses the underlying science, developmental data, and operational context that supports the designation of ECs. This includes detailed facility blueprints, validation master plans, general operating standard operating procedures (SOPs), and experimental data from early pilot scales. Modifications to sustaining information do not require regulatory notification and are managed entirely via internal site change control protocols.
Post-Approval Change Management Procedures (PACMPs)
A PACMP is a comprehensive, legally binding plan that details a specific manufacturing modification that the sponsor intends to implement throughout the product lifecycle. The protocol explicitly outlines:
The exact nature of the proposed modifications (such as changing an analytical method, upgrading a bioreactor configuration, or transferring an active pharmaceutical ingredient to an alternate facility).
The risk-management strategy is used to evaluate the possible impact of the modification on product quality attributes.
The specific analytical testing, validation matrix, and stability commitments are required to show product comparability.
The predetermined down-regulated reporting category (for example, converting what would typically be a prior-approval Supplement into a post-implementation Notification) if all specified acceptance criteria are met.
By securing prior agency approval for the testing methodology and downgrading logic in the initial PACMP submission, manufacturers can implement modifications quickly once internal testing confirms success.
Structural Requirements of a Mature Pharmaceutical Quality System (PQS)
Sponsors cannot utilize the regulatory flexibilities of ICH Q12 without showing a functional, highly capable PQS that complies with ICH Q10 principles. Regulatory bodies will not grant down-regulated change pathways to facilities lacking robust, data-driven internal quality governance.
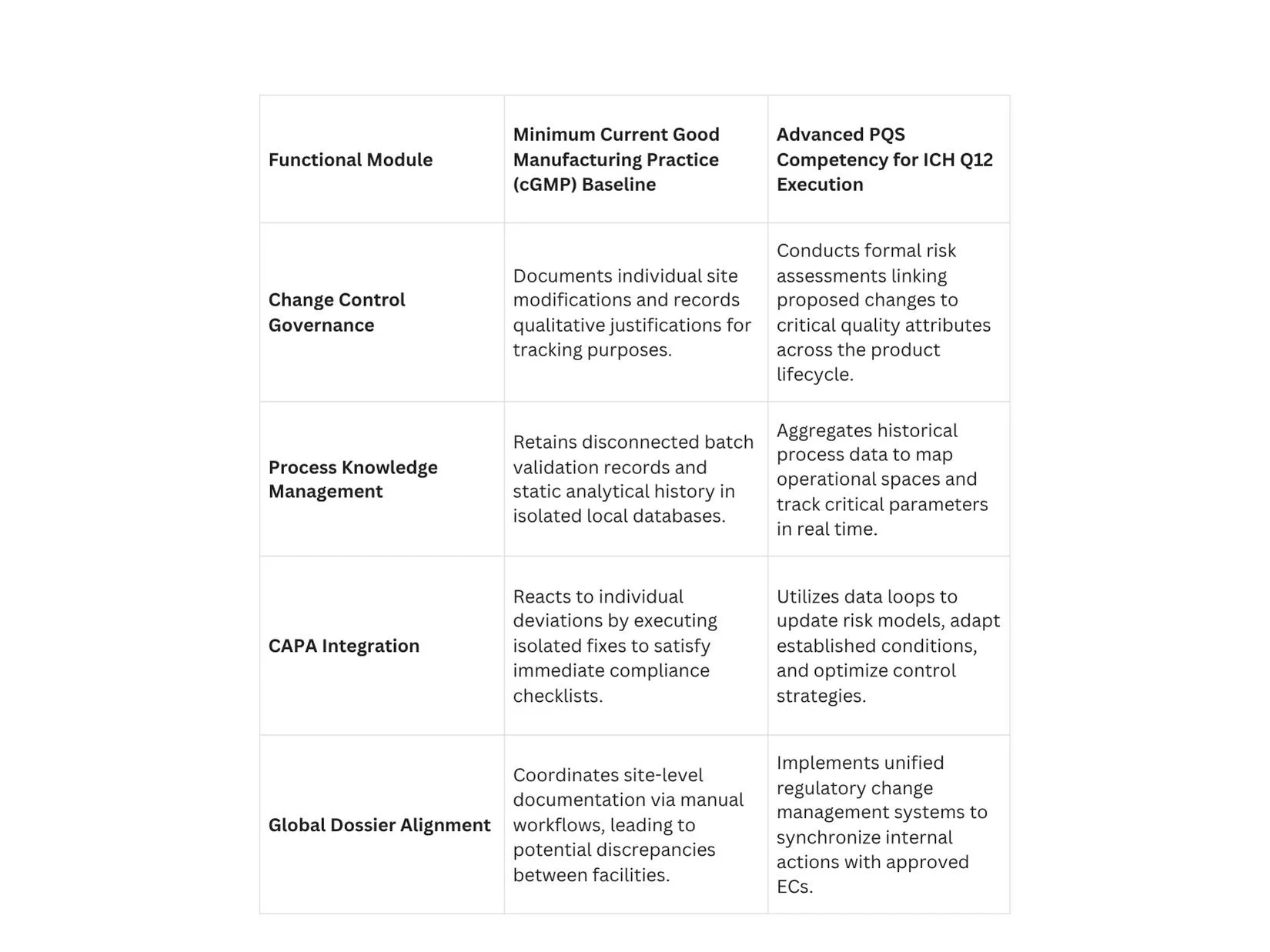
Process Knowledge Management across the Lifecycle
A compliant PQS must operate a continuous knowledge management framework that captures data from early clinical development through commercial manufacturing. Process knowledge should not be stored in isolated paper batch records or disparate local databases.
Instead, it must be aggregated into unified data structures that clearly reflect process parameters, material sources, and environmental variables. This deep process knowledge provides the scientific basis for proposing, justifying, and defending specific boundaries for Established Conditions during regulatory audits.
Regulatory Reporting Categories and Downgrading Strategies
The implementation of ICH Q12 provides a mechanism to modify the default regulatory reporting structures defined by regional laws. The goal is to move low-risk, well-understood adjustments out of prior-approval queues and into post-implementation notification pathways.
The Mechanism of Risk-Based Downgrading
When a manufacturer demonstrates an extensive understanding of a process, they can propose a risk-based categorization strategy for individual ECs. For instance, a process parameter with a broad operating margin and minimal impact on structural attributes can be negotiated from a major reporting tier down to a minor tier.
When this strategy is combined with an approved PACMP, the process efficiency increases significantly. A site transfer for a complex biologic that traditionally required a detailed, prior-approval application can be executed as a post-notice change, provided the verification data satisfies the criteria defined in the protocol.
Introduction of Immediate Notification Pathways
To support such flexibility, modernized regulatory revisions are launching new communication mechanisms. For example, Health Canada introduced a Level III Immediate Notification category within its updated framework. This reporting tier accommodates modifications that have been downgraded from higher risk categories via approved ICH Q12 enablers.
Sponsors utilizing this pathway must notify the agency within 15 days of releasing the modified product to the Canadian market, allowing the regulatory body to maintain oversight without delaying commercial supply lines.
Technical Step-by-Step Implementation Protocol for PACMP Execution
Successfully executing an approved PACMP requires strict adherence to a systematic operational workflow to preserve compliance throughout the product lifecycle.
Phase 1: Protocol Development and Submission
The sponsor prepares a comprehensive PACMP submission within the initial marketing authorization application or via a subsequent formal variation supplement. This document must include a precise description of the future change, the risk mitigation strategy, the analytical methods to be used, and the targeted down-regulated reporting category. The protocol must be reviewed and approved by the target regulatory authority before any subsequent lifecycle modifications can use this pathway.
Phase 2: Internal Facility Execution and Validation
Once the protocol is approved, the manufacturer can initiate the physical change at the designated facility. For example, if transferring production to a new manufacturing line, the site must install the equipment, execute installation and operational qualifications, and run commercial-scale comparison batches.
All analytical data, validation outputs, and stability testing must be conducted exactly as specified in the approved PACMP.
Phase 3: Data Comparison and Acceptance Verification
The quality unit compiles the resulting analytical data and evaluates it against the predetermined acceptance criteria established in the protocol.
If all parameters fall within the approved boundaries, the change is considered successful. If any metric fails to meet the criteria, the protocol becomes invalid for that modification, and the change must revert to the standard prior-approval submission pathway.
Phase 4: Post-Implementation Reporting
Upon verifying compliance, the manufacturer implements the change in commercial production. The sponsor then files the required regulatory notice under the agreed-down-regulated category, such as an immediate notification or an annual report, citing the approved protocol reference number and providing the supporting validation data package.
Commercial and Operational Impact on Global Supply Chains
The transition to an ICH Q12 framework delivers significant strategic and commercial gains that extend beyond basic regulatory compliance.
Mitigating Drug Shortages through Agility
A primary driver of ICH Q12 adoption is the prevention of pharmaceutical supply interruptions and critical drug shortages. In traditional regulatory systems, expanding manufacturing capacity or onboarding an alternative raw-material supplier could take months due to backlogs in prior-approval queues.
By using approved PACMPs and clearly delineated ECs, manufacturers can activate backup manufacturing facilities and alternative material pipelines within days. This nimbleness secures a continuous supply of critical therapeutics to global markets.
Accelerating Ongoing Enhancement and Innovation
The traditional oversight model inadvertently penalized innovation by mandating extensive regulatory filings for minor process improvements. This administrative burden frequently led manufacturers to run outdated processes rather than handle the complex post-approval review landscape.
ICH Q12 removes these barriers, enabling companies to continuously optimize production lines, implement real-time release testing, and deploy advanced process analytical technologies (PAT) under internal PQS controls. This continuous optimization drives lower operating expenses, reduces batch failure rates, and increases overall manufacturing yields.
Conclusion: The Strategic Criticality of Operationalizing ICH Q12
The implementation of ICH Q12 marks a fundamental shift toward an optimized, data-driven approach to pharmaceutical lifecycle management. By implementing tools such as Post-Approval Change Management Procedures and explicitly mapped Established Conditions, manufacturers can significantly reduce regulatory timelines and accelerate facility upgrades.
However, these advanced regulatory flexibilities cannot operate in a vacuum; they require a highly capable, data-driven Pharmaceutical Quality System built on robust knowledge management and risk-based decision-making. As regulatory authorities globally continue to embed these guidelines into their standard oversight frameworks, companies that fail to operationalize these enablers risk a permanent competitive and operational disadvantage.
To review the scientific consensus, emerging clinical data, and peer-reviewed case studies supporting advanced lifecycle management strategies, quality professionals can access Nature.com to ensure their operational systems comply with current international practices.
Don't let rigid regulatory frameworks hold back your manufacturing innovation or compromise your supply chain stability. Contact Metis Consulting Services today.