
ICH Q12: Post-Approval Change Management for Pharmaceutical Product Lifecycle Management
This week in the Guardrail, we break down the practical mechanics of the ICH Q12 framework. Tools like Established Conditions and Post-Approval Change Management Procedures can streamline regulatory paths and protect global supply chains.
By Michael Bronfman
May 29, 2026
The global pharmaceutical regulatory framework is transitioning from a rigid, reactive paradigm to an anticipatory, science and risk-based lifecycle management model. Central to this transformation remains the international implementation of the International Council for Harmonization (ICH) Q12 guideline.
Historically, post-approval changes (PACs) to chemistry, manufacturing, and controls (CMC) required extensive, multi-jurisdictional regulatory reviews. These extended processes frequently delayed the introduction of manufacturing innovations, equipment upgrades, and site transfers.
The formalized roll-out of ICH Q12 mechanisms introduces an organized approach to identifying and managing regulatory commitments. This framework allows manufacturers to execute routine modifications under the oversight of their internal Pharmaceutical Quality System (PQS), reducing the burden of prior-approval regulatory filings.
Evolving Jurisdictional Implementation Boundaries
Global regulatory bodies are adopting the tools and enablers outlined in ICH Q12 at varying paces and within specific product domains.
Health Canada Strategy
Health Canada has introduced updates to its regulatory infrastructure, denoting a step-wise integration of the ICH Q12 framework. The Biologic and Radiopharmaceutical Drugs Directorate (BRDD) updated its Health Canada Guidance on Post-Notice of Compliance Changes Framework to establish the operational boundaries for these tools.
Initial implementation focuses exclusively on Post-Approval Change Management Procedures (PACMPs) for products regulated by the BRDD, including biologics and Schedule C drugs. Under this system, the submission of qualifying PACMPs will be formally accepted following a 90-day transition period ending August 13, 2026.
Notably, Established Conditions (ECs) for all product classes and PACMPs for applications outside the BRDD fall outside the initial scope. Broader integration by the Pharmaceutical Drugs Directorate (PDD) is scheduled for subsequent phases, with particular timelines expected later in the year.
Global Agency Status
The United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) provide precedents for these tools across chemical entities and therapeutic biologics. These agencies accept regulatory submissions containing explicitly defined ECs and PACMPs, provided the manufacturer demonstrates an advanced PQS during routine facility inspections.
The differences in implementation speed underscore the need for multinational pharmaceutical operations to design global change strategies that navigate different regulatory requirements.
Functional Mechanics of ICH Q12 Regulatory Tools
The practical value of ICH Q12 relies on two interconnected instruments: Established Conditions and Post-Approval Change Management Guidelines. These tools shift the focus of regulatory dossiers from arbitrary data elements to critical-to-quality variables.
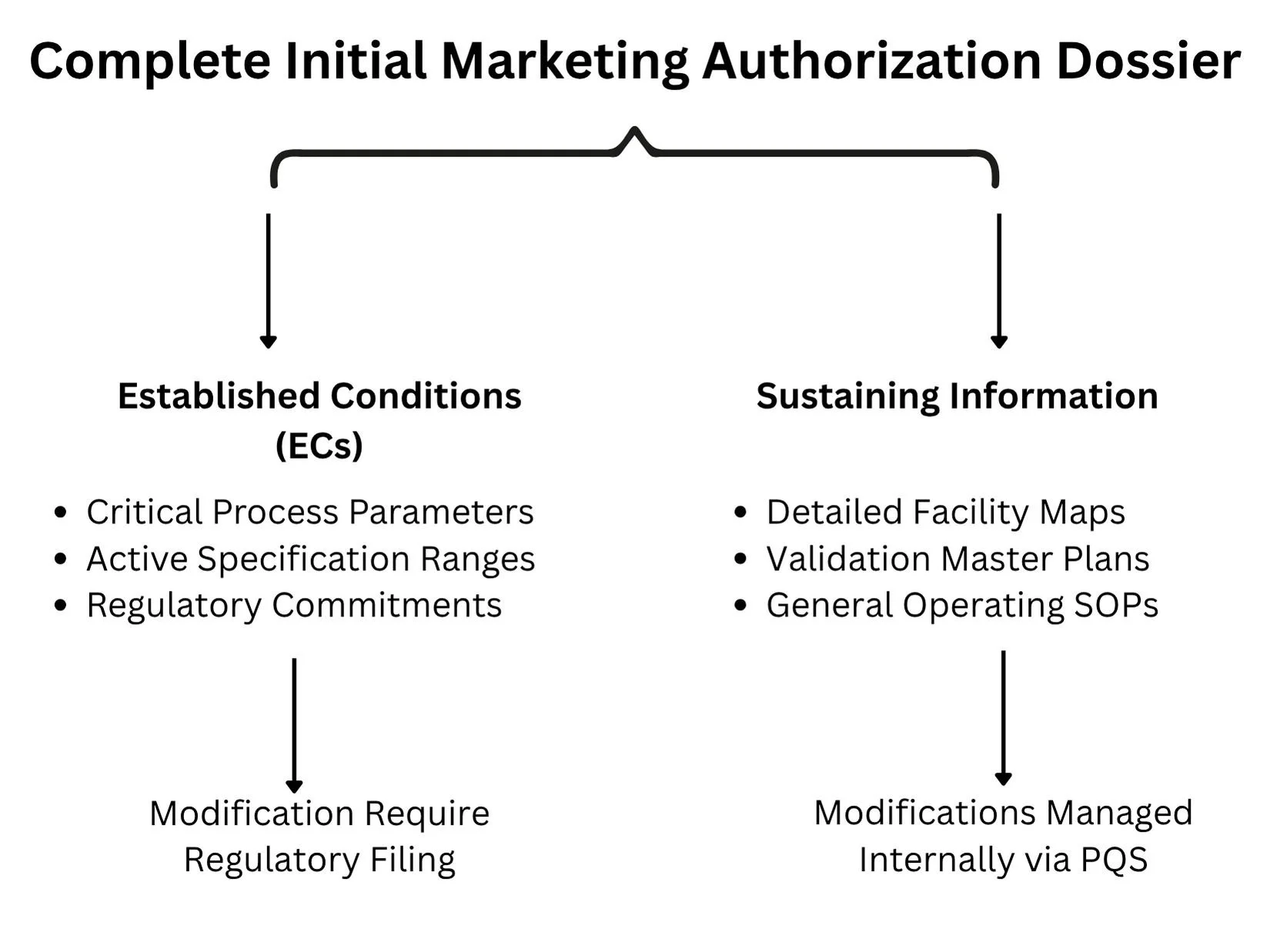
Delineating Established Conditions from Sustaining Information
A longstanding challenge in lifecycle management has been the lack of a clear distinction between legally binding regulatory commitments and purely illustrative descriptive text in the Common Technical Document (CTD). ICH Q12 tackles this by separating information into Established Conditions and Sustaining Information.
Established Conditions (ECs): Defined as the specific elements of a manufacture or control strategy necessary to ensure product safety, identity, strength, purity, or potency. Any modification to an approved EC constitutes a regulatory change that must be reported to the oversight agency. Examples include critical process parameters (CPPs), critical quality attributes (CQAs), acceptance criteria for raw materials, and active operational dimensions of specialized purification columns.
Sustaining Information: Encompasses the underlying science, developmental data, and operational context that supports the designation of ECs. This includes detailed facility blueprints, validation master plans, general operating standard operating procedures (SOPs), and experimental data from early pilot scales. Modifications to sustaining information do not require regulatory notification and are managed entirely via internal site change control protocols.
Post-Approval Change Management Procedures (PACMPs)
A PACMP is a comprehensive, legally binding plan that details a specific manufacturing modification that the sponsor intends to implement throughout the product lifecycle. The protocol explicitly outlines:
The exact nature of the proposed modifications (such as changing an analytical method, upgrading a bioreactor configuration, or transferring an active pharmaceutical ingredient to an alternate facility).
The risk-management strategy is used to evaluate the possible impact of the modification on product quality attributes.
The specific analytical testing, validation matrix, and stability commitments are required to show product comparability.
The predetermined down-regulated reporting category (for example, converting what would typically be a prior-approval Supplement into a post-implementation Notification) if all specified acceptance criteria are met.
By securing prior agency approval for the testing methodology and downgrading logic in the initial PACMP submission, manufacturers can implement modifications quickly once internal testing confirms success.
Structural Requirements of a Mature Pharmaceutical Quality System (PQS)
Sponsors cannot utilize the regulatory flexibilities of ICH Q12 without showing a functional, highly capable PQS that complies with ICH Q10 principles. Regulatory bodies will not grant down-regulated change pathways to facilities lacking robust, data-driven internal quality governance.
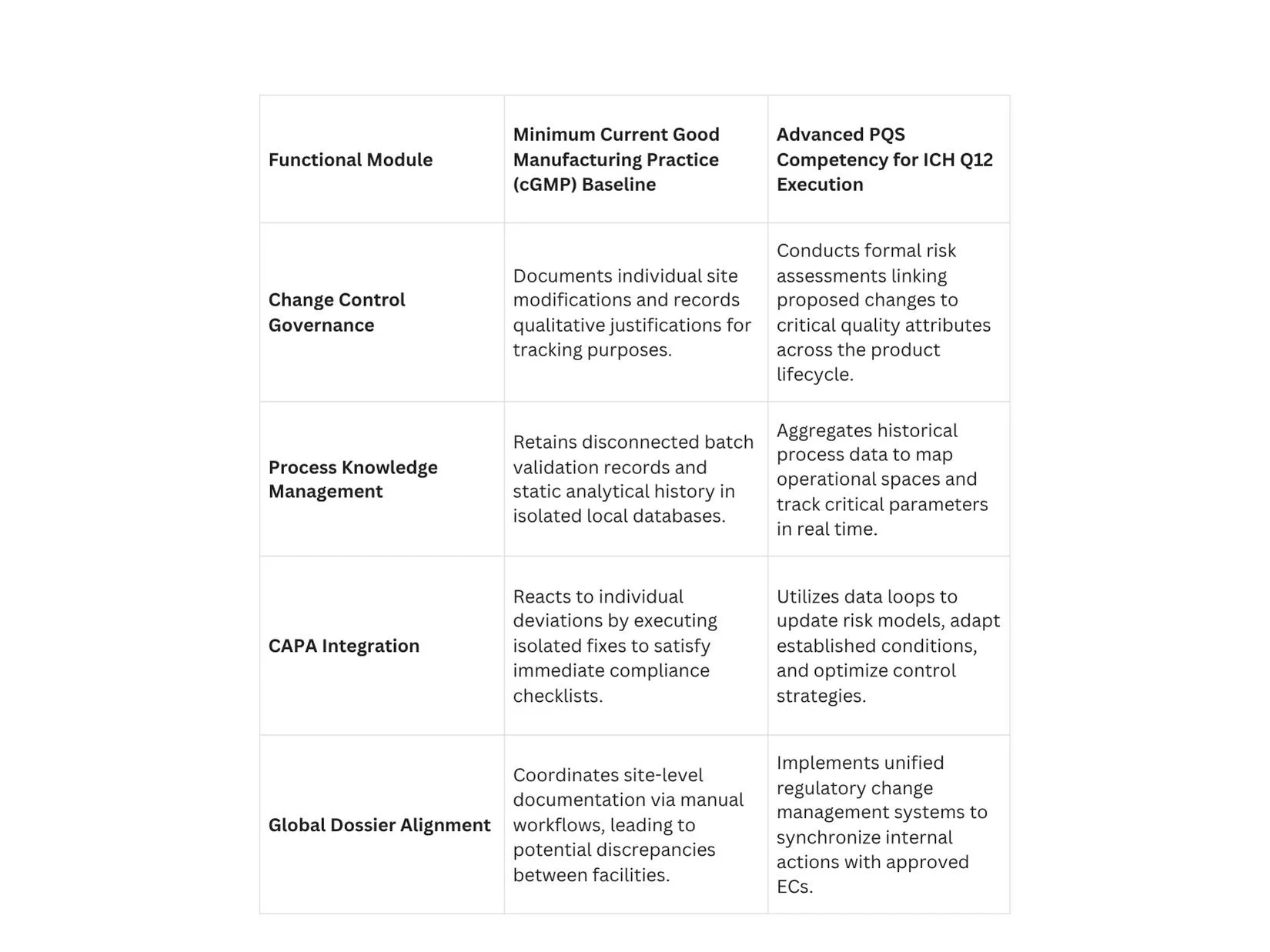
Process Knowledge Management across the Lifecycle
A compliant PQS must operate a continuous knowledge management framework that captures data from early clinical development through commercial manufacturing. Process knowledge should not be stored in isolated paper batch records or disparate local databases.
Instead, it must be aggregated into unified data structures that clearly reflect process parameters, material sources, and environmental variables. This deep process knowledge provides the scientific basis for proposing, justifying, and defending specific boundaries for Established Conditions during regulatory audits.
Regulatory Reporting Categories and Downgrading Strategies
The implementation of ICH Q12 provides a mechanism to modify the default regulatory reporting structures defined by regional laws. The goal is to move low-risk, well-understood adjustments out of prior-approval queues and into post-implementation notification pathways.
The Mechanism of Risk-Based Downgrading
When a manufacturer demonstrates an extensive understanding of a process, they can propose a risk-based categorization strategy for individual ECs. For instance, a process parameter with a broad operating margin and minimal impact on structural attributes can be negotiated from a major reporting tier down to a minor tier.
When this strategy is combined with an approved PACMP, the process efficiency increases significantly. A site transfer for a complex biologic that traditionally required a detailed, prior-approval application can be executed as a post-notice change, provided the verification data satisfies the criteria defined in the protocol.
Introduction of Immediate Notification Pathways
To support such flexibility, modernized regulatory revisions are launching new communication mechanisms. For example, Health Canada introduced a Level III Immediate Notification category within its updated framework. This reporting tier accommodates modifications that have been downgraded from higher risk categories via approved ICH Q12 enablers.
Sponsors utilizing this pathway must notify the agency within 15 days of releasing the modified product to the Canadian market, allowing the regulatory body to maintain oversight without delaying commercial supply lines.
Technical Step-by-Step Implementation Protocol for PACMP Execution
Successfully executing an approved PACMP requires strict adherence to a systematic operational workflow to preserve compliance throughout the product lifecycle.
Phase 1: Protocol Development and Submission
The sponsor prepares a comprehensive PACMP submission within the initial marketing authorization application or via a subsequent formal variation supplement. This document must include a precise description of the future change, the risk mitigation strategy, the analytical methods to be used, and the targeted down-regulated reporting category. The protocol must be reviewed and approved by the target regulatory authority before any subsequent lifecycle modifications can use this pathway.
Phase 2: Internal Facility Execution and Validation
Once the protocol is approved, the manufacturer can initiate the physical change at the designated facility. For example, if transferring production to a new manufacturing line, the site must install the equipment, execute installation and operational qualifications, and run commercial-scale comparison batches.
All analytical data, validation outputs, and stability testing must be conducted exactly as specified in the approved PACMP.
Phase 3: Data Comparison and Acceptance Verification
The quality unit compiles the resulting analytical data and evaluates it against the predetermined acceptance criteria established in the protocol.
If all parameters fall within the approved boundaries, the change is considered successful. If any metric fails to meet the criteria, the protocol becomes invalid for that modification, and the change must revert to the standard prior-approval submission pathway.
Phase 4: Post-Implementation Reporting
Upon verifying compliance, the manufacturer implements the change in commercial production. The sponsor then files the required regulatory notice under the agreed-down-regulated category, such as an immediate notification or an annual report, citing the approved protocol reference number and providing the supporting validation data package.
Commercial and Operational Impact on Global Supply Chains
The transition to an ICH Q12 framework delivers significant strategic and commercial gains that extend beyond basic regulatory compliance.
Mitigating Drug Shortages through Agility
A primary driver of ICH Q12 adoption is the prevention of pharmaceutical supply interruptions and critical drug shortages. In traditional regulatory systems, expanding manufacturing capacity or onboarding an alternative raw-material supplier could take months due to backlogs in prior-approval queues.
By using approved PACMPs and clearly delineated ECs, manufacturers can activate backup manufacturing facilities and alternative material pipelines within days. This nimbleness secures a continuous supply of critical therapeutics to global markets.
Accelerating Ongoing Enhancement and Innovation
The traditional oversight model inadvertently penalized innovation by mandating extensive regulatory filings for minor process improvements. This administrative burden frequently led manufacturers to run outdated processes rather than handle the complex post-approval review landscape.
ICH Q12 removes these barriers, enabling companies to continuously optimize production lines, implement real-time release testing, and deploy advanced process analytical technologies (PAT) under internal PQS controls. This continuous optimization drives lower operating expenses, reduces batch failure rates, and increases overall manufacturing yields.
Conclusion: The Strategic Criticality of Operationalizing ICH Q12
The implementation of ICH Q12 marks a fundamental shift toward an optimized, data-driven approach to pharmaceutical lifecycle management. By implementing tools such as Post-Approval Change Management Procedures and explicitly mapped Established Conditions, manufacturers can significantly reduce regulatory timelines and accelerate facility upgrades.
However, these advanced regulatory flexibilities cannot operate in a vacuum; they require a highly capable, data-driven Pharmaceutical Quality System built on robust knowledge management and risk-based decision-making. As regulatory authorities globally continue to embed these guidelines into their standard oversight frameworks, companies that fail to operationalize these enablers risk a permanent competitive and operational disadvantage.
To review the scientific consensus, emerging clinical data, and peer-reviewed case studies supporting advanced lifecycle management strategies, quality professionals can access Nature.com to ensure their operational systems comply with current international practices.
Don't let rigid regulatory frameworks hold back your manufacturing innovation or compromise your supply chain stability. Contact Metis Consulting Services today.